
Visi iLive saturs ir medicīniski pārskatīts vai pārbaudīts, lai nodrošinātu pēc iespējas lielāku faktisko precizitāti.
Mums ir stingras iegādes vadlīnijas un tikai saikne ar cienījamiem mediju portāliem, akadēmiskām pētniecības iestādēm un, ja vien iespējams, medicīniski salīdzinošiem pārskatiem. Ņemiet vērā, ka iekavās ([1], [2] uc) esošie numuri ir klikšķi uz šīm studijām.
Ja uzskatāt, ka kāds no mūsu saturiem ir neprecīzs, novecojis vai citādi apšaubāms, lūdzu, atlasiet to un nospiediet Ctrl + Enter.
Achondroplāzija
Raksta medicīnas eksperts

Pēdējā pārskatīšana: 04.07.2025
Pastāv daudzas retas iedzimtas slimības, un viena no tām ir kaulu augšanas pārkāpums - ahondroplāzija, kas noved pie smagas nesamērīgas maza auguma.
ICD-10 sadaļā par attīstības anomālijām šāda veida iedzimtas osteohondrālas displāzijas ar cauruļveida kaulu un mugurkaula augšanas defektiem kods ir Q77.4 [ 1 ]
Epidemioloģija
Runājot par ahondroplāzijas izplatību, dažādu pētījumu statistikas dati ir neviennozīmīgi. Daži apgalvo, ka šī anomālija rodas vienam jaundzimušajam no 10 tūkstošiem, citi - vienam no 26-28 tūkstošiem, bet vēl citi - 4-15 gadījumos no 100 tūkstošiem. [ 2 ]
Ir arī informācija, ka, ja tēvs ir vecāks par 50 gadiem, ahondroplāzijas sastopamība bērniem ir viens gadījums uz 1875 jaundzimušajiem.
Cēloņi ahondroplāzija
Ahondroplāzijas cēlonis ir osteoģenēzes pārkāpums, jo īpaši viens no skeleta cauruļveida kaulu diafīzes intrauterīnās osifikācijas veidiem - endohondrālā osifikācija, kuras laikā skrimšļi tiek modificēti kaulu audos. Sīkāku informāciju skatiet sadaļā - Kaulu attīstība un augšana.
Garo kaulu osifikācijas traucējumi jeb augļa ahondroplāzija rodas membrānas tirozīnkināzes gēna - fibroblastu augšanas faktora receptora 3 (FGFR3 4p16.3 hromosomā) mutāciju dēļ, kas ietekmē šūnu augšanu un diferenciāciju. FGFR3 mutāciju klātbūtne ir saistīta ar ģenētisku nestabilitāti un hromosomu skaita izmaiņām (aneiploīdiju).
Ahondroplāzija bērnam tiek pārmantota kā autosomāli dominējoša pazīme, tas ir, viņš saņem vienu mutanta gēna kopiju (kas ir dominējošais) un vienu normālu gēnu uz nedzimuma (autosomālu) hromosomu pāra. Tādējādi šī defekta mantojuma veids ir autosomāli dominējošais, un anomālija var izpausties 50% pēcnācēju, ja tiek krustota šī gēna (genotipa) alēļu kombinācija.
Turklāt mutācijas var būt sporādiskas, un, kā liecina prakse, 80% gadījumu bērni ar ahondroplāziju piedzimst vecākiem ar normālu augumu.
Riska faktori
Galvenie riska faktori bērnu ar ahondroplāziju piedzimšanai ir iedzimtība. Ja vienam no vecākiem ir šis defekts, tad slima bērna piedzimšanas varbūtība tiek lēsta 50% apmērā; ja abiem vecākiem ir šī anomālija, tā ir arī 50%, bet ar 25% homozigotas ahondroplāzijas risku, kas noved pie nāves pirms dzimšanas vai agrā zīdaiņa vecumā.
Pieaugot tēva vecumam (tuvāk 40 gadiem un vecākiem), palielinās FGFR3 gēna jaunas mutācijas (de novo mutācijas) risks.
Pathogenesis
Skaidrojot ahondroplāzijas patoģenēzi, eksperti uzsver transmembrānas proteīna tirozīnproteīnkināzes (ko kodē FGFR3 gēns) nozīmi augšanas plātnīšu skrimšļa audu šūnu - hondrocītu - dalīšanās, diferenciācijas un apoptozes regulēšanā, kā arī skeleta normālas attīstības - osteoģenēzes un kaulu audu mineralizācijas - norisē.
Embrionālās attīstības laikā, gēnu mutācijas klātbūtnē, fibroblastu augšanas faktora 3 receptori kļūst aktīvāki. To funkciju palielināšanās traucē šūnu signālu pārraidi un šī proteīna ārpusšūnu daļas mijiedarbību ar polipeptīdu fibroblastu augšanas faktoriem (FGF). Rezultātā rodas kļūme: skrimšļu šūnu proliferācijas stadija kļūst īsāka, un to diferenciācija sākas agrāk nekā paredzēts. Tas viss noved pie galvaskausa kaulu nepareizas veidošanās un saplūšanas un skeleta displāzijas - garo kaulu samazināšanās, ko pavada izteikts mazs augums jeb pundurisms.
Un divas trešdaļas pundurisma gadījumu ir saistīti ar ahondroplāziju.
Simptomi ahondroplāzija
Patoloģiska kaulu augšana izraisa ahondroplāzijas klīniskos simptomus, piemēram:
- izteikts īss augums (nesamērīgs pundurisms) ar vidējo pieaugušā cilvēka augumu 123–134 cm;
- apakšējo un augšējo ekstremitāšu proksimālo daļu saīsināšana ar relatīvi normālu rumpja izmēru;
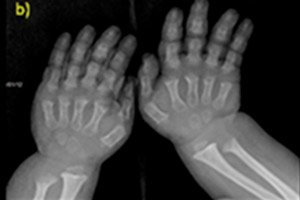
- saīsināti pirkstu un kāju pirksti;
- palielināta galva (makro vai megalocefālija); [ 3 ]
- specifiskas sejas iezīmes izvirzītas pieres un sejas vidusdaļas hipoplāzijas veidā - iegrimis deguna tilts.
- Šaura galvaskausa un kakla savienojuma vieta. Daži zīdaiņi ar ahondroplāziju mirst pirmajā dzīves gadā no komplikācijām, kas saistītas ar galvaskausa un kakla savienojumu; populācijas pētījumi liecina, ka bez novērtēšanas un iejaukšanās šis pārmērīgais nāves risks var sasniegt pat 7,5 %.[ 4 ]
- Vidusauss disfunkcija bieži ir problēma [ 5 ], un, ja to neārstē pareizi, tā var izraisīt vadītspējīgu dzirdes zudumu, kas ir pietiekami smags, lai traucētu runas attīstību. Vairāk nekā pusei bērnu būs nepieciešama spiediena izlīdzināšanas caurule. [ 6 ] Kopumā aptuveni 40% cilvēku ar ahondroplāziju ir funkcionāli nozīmīgs dzirdes zudums. Arī ekspresīvās valodas attīstība bieži vien ir aizkavēta, lai gan saiknes stiprums starp dzirdes zudumu un ekspresīvās valodas problēmām ir apšaubāms.
- Lielā stilba kaula izliekums ir ļoti izplatīts cilvēkiem ar ahondroplāziju. Vairāk nekā 90 % neārstētu pieaugušo ir zināmā mērā izliekts.[ 7 ] "Izliekums" patiesībā ir sarežģīta deformācija, kas rodas sānu noliekuma, liela kaula iekšējās vērpes un ceļa locītavas dinamiskās nestabilitātes kombinācijas rezultātā.[ 8 ]
Zīdaiņiem ar ahondroplāziju raksturīga muskuļu hipotonija, kuras dēļ viņi sāk apgūt kustību prasmes un staigāt vēlāk. Šis attīstības defekts neietekmē intelektu un kognitīvās spējas. [ 9 ], [ 10 ]
Sekas un komplikācijas
Šāda veida iedzimtu osteohondrālu displāziju raksturo šādas komplikācijas un sekas:
- atkārtotas ausu infekcijas;
- obstruktīva miega apnoja;
- hidrocefālija;
- maloklūzija un šķībi zobi:
- kāju deformācija (varus vai valgus) ar gaitas izmaiņām;
- jostas daļas hipertrofēta lordoze vai tās izliekums (krūškurvja un jostas daļas kifoze vai jostas daļas skolioze) - ar muguras sāpēm ejot;
- locītavu sāpes (nepareizas kaulu novietojuma vai nervu saknīšu saspiešanas dēļ);
- Mugurkaula stenoze un muguras smadzeņu kompresija; Visbiežāk sastopamā medicīniskā sūdzība pieaugušo vecumā ir simptomātiska mugurkaula stenoze, kas skar L1-L4. Simptomi ir no intermitējošas, atgriezeniskas klibošanas, ko izraisa fiziska slodze, līdz smagai, neatgriezeniskai kāju disfunkcijai un urīna aizturei.[ 11 ] Klibošana un stenoze var izraisīt gan sensorus (nejutīgums, sāpes, smagums), gan motoriskus simptomus (vājums, klupšana, ierobežota iešanas izturība). Vaskulārā klibošana rodas asinsvadu pietūkuma rezultātā pēc stāvēšanas un staigāšanas, un tā ir pilnībā atgriezeniska ar atpūtu. Mugurkaula stenoze ir muguras smadzeņu vai nervu saknītes faktisks bojājums, ko izraisa mugurkaula kanāla stenozētais kauls, un simptomi ir neatgriezeniski. Simptomi, kas lokalizējas konkrētā dermatomā, var rasties specifisku nervu saknītes atveru stenozes dēļ.
- krūškurvja sienas samazināšanās ar ierobežotu plaušu augšanu un samazinātu plaušu funkciju (smaga elpas trūkums). Zīdaiņa vecumā nelielai cilvēku grupai ar ahondroplāziju ir restriktīvas plaušu problēmas. Mazas krūtis un palielināta krūškurvja elastība kopā izraisa samazinātu plaušu tilpumu un restriktīvu plaušu slimību [ 12 ].
Citas ortopēdiskas problēmas
- Locītavu vājums. Lielākā daļa locītavu bērnībā ir hipermobilas. Parasti tam ir maza ietekme, izņemot ceļa nestabilitāti dažiem cilvēkiem.
- Diskveida laterālais menisks: šī nesen atklātā strukturālā anomālija dažiem cilvēkiem var izraisīt hroniskas ceļa sāpes.[ 13 ]
- Artrīts: FGFR-3 konstitutīva aktivācija, tāpat kā ahondroplāzijas gadījumā, var pasargāt no artrīta attīstības.[ 14 ]
- Acanthosis nigricans ir novērojama aptuveni 10% cilvēku ar ahondroplāziju.[ 15 ] Šajā populācijā tā neliecina par hiperinsulinēmiju vai ļaundabīgu audzēju.
Homozigota ahondroplāzija, ko izraisa bialēļu patogēni varianti FGFR3 1138. nukleotīdā, ir smaga slimība ar radioloģiskiem atradumiem, kas kvalitatīvi atšķiras no tiem, kas novēroti ahondroplāzijas gadījumā. Agrīna nāve rodas elpošanas mazspējas dēļ, ko izraisa šaura krūškurvja siena, un neiroloģisku deficītu, ko izraisa cervikomedulāra stenoze [Hall 1988].
Diagnostika ahondroplāzija
Vairumam pacientu ahondroplāzijas diagnoze tiek noteikta, pamatojoties uz raksturīgām klīniskām pazīmēm un radiogrāfiskiem atradumiem. Zīdaiņiem vai dažu simptomu neesamības gadījumā galīgās diagnozes noteikšanai tiek izmantoti ģenētiskie testi, piemēram, kariotipa analīze.[16 ]
Veicot pirmsdzemdību diagnostiku, izmantojot molekulārās ģenētikas metodi, var veikt augļūdeņu vai horiona bārkstiņu parauga analīzes.
Ahondroplāzijas pazīmes augļa ultraskaņā - ekstremitāšu saīsināšanās un tipiskas sejas iezīmes - tiek vizualizētas pēc 22 grūtniecības nedēļām.
Instrumentālā diagnostika ietver arī skeleta rentgenu vai kaulu ultraskaņu. Rentgens apstiprina diagnozi, pamatojoties uz tādiem datiem kā liels galvaskauss ar šauru pakauša atveri un relatīvi mazu pamatni; īsi cauruļkauli un saīsinātas ribas; īsi un saplacināti skriemeļu ķermeņi; sašaurināts mugurkaula kanāls, samazināts iegurņa kaula spārnu izmērs.
Diferenciālā diagnoze
Nepieciešama diferenciāldiagnostika ar hipofīzes pundurismu, iedzimtu spondiloepifizāru un diastrofisku displāziju, hipohondroplāziju, Šereševska-Tērnera un Nūnana sindromiem, pseidoahondroplāziju. Tādējādi atšķirība starp pseidoahondroplāziju un ahondroplāziju ir tāda, ka pacientiem ar pundurismu pseidoahondroplāzijas gadījumā galvas izmērs un sejas vaibsti ir normāli.
Kurš sazināties?
Profilakse
Vienīgais preventīvais pasākums ir iedzimtu slimību pirmsdzemdību diagnostika. [ 41 ], [ 42 ]
Prognoze
Cik ilgi dzīvo cilvēki ar ahondroplāziju? Apmēram par 10 gadiem īsāk nekā vidējais paredzamais dzīves ilgums.
Tā kā patoloģiskas izmaiņas kaulu audos un locītavās ierobežo pašaprūpes un mobilitāti, bērniem ar šo diagnozi tiek piešķirts invalīda statuss. Ilgtermiņā lielākajai daļai pacientu ir normāla prognoze, taču ar vecumu palielinās sirds slimību risks. [ 43 ]