
Visi iLive saturs ir medicīniski pārskatīts vai pārbaudīts, lai nodrošinātu pēc iespējas lielāku faktisko precizitāti.
Mums ir stingras iegādes vadlīnijas un tikai saikne ar cienījamiem mediju portāliem, akadēmiskām pētniecības iestādēm un, ja vien iespējams, medicīniski salīdzinošiem pārskatiem. Ņemiet vērā, ka iekavās ([1], [2] uc) esošie numuri ir klikšķi uz šīm studijām.
Ja uzskatāt, ka kāds no mūsu saturiem ir neprecīzs, novecojis vai citādi apšaubāms, lūdzu, atlasiet to un nospiediet Ctrl + Enter.
Šarko Marijas-Toota slimība.
Raksta medicīnas eksperts

Pēdējā pārskatīšana: 04.07.2025
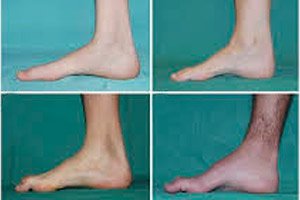
Peroneālā muskuļu atrofija, Šarko-Marijas-Touta sindroms vai slimība ir hronisku iedzimtu slimību grupa, kas izraisa perifēro nervu bojājumus.
Saskaņā ar ICD-10, sadaļā par nervu sistēmas slimībām šīs slimības kods ir G60.0 (iedzimta motorā un sensorā neiropātija). Tā ir iekļauta arī bāreņu slimību sarakstā.
Epidemioloģija
Saskaņā ar klīnisko statistiku, visu veidu Šarko-Marī-Toota slimības izplatība uz 100 tūkstošiem iedzīvotāju ir 19 gadījumi (saskaņā ar citiem avotiem, viens gadījums uz 2,5–10 tūkstošiem iedzīvotāju).
CMT 1. tips veido aptuveni divas trešdaļas gadījumu (viens gadījums uz 5000–7000 iedzīvotāju), un gandrīz 70 % no tiem ir saistīti ar PMP22 gēna dublēšanos. No šāda veida slimības visā pasaulē cieš vairāk nekā 1,2 miljoni cilvēku.
CMT 4. tipa sastopamība tiek lēsta 1–5 gadījumos uz 10 000 bērniem. [ 1 ]
Cēloņi Šarko Marijas-Toota slimība
Saskaņā ar polineiropātijas sindromu klasifikāciju peroneālā (fibulārā) muskuļu atrofija, Šarko-Marijas-Tota neirālā amiotrofija vai Šarko-Marijas-Tota slimība (saīsināti CMT) attiecas uz ģenētiski determinētām motoriski sensorām polineiropātijām. [ 2 ]
Tas nozīmē, ka tā rašanās cēloņi ir ģenētiskas mutācijas. Un atkarībā no ģenētisko noviržu rakstura izšķir galvenos šī sindroma veidus vai veidus: demielinizējošu un aksonālu. Pirmajā grupā ietilpst 1. tipa Šarko-Marijas-Tota slimība (CMT1), kas rodas PMP22 gēna dublēšanās dēļ 17. hromosomā, kas kodē transmembrānu perifēro mielīna proteīnu 22. Tā rezultātā notiek aksona apvalka (nervu šūnu procesu) segmentāla demielinizācija un nervu signāla vadīšanas ātruma samazināšanās. Turklāt mutācijas var būt arī dažos citos gēnos.
Aksonālā forma ir Šarko-Marijas-Tootas slimības 2. tips (CMT2), kas ietekmē pašus aksonus un ir saistīta ar patoloģiskām izmaiņām MFN2 gēnā lokusā 1p36.22, kas kodē membrānas proteīnu mitofuzīnu-2, kas ir nepieciešams mitohondriju saplūšanai un funkcionālu mitohondriju tīklu veidošanai perifēro nervu šūnās. Pastāv vairāk nekā ducis CMT2 apakštipu (ar mutācijām specifiskos gēnos).
Jāatzīmē, ka pašlaik ir identificēti vairāk nekā simts gēnu, kuru bojājumi, kas tiek pārmantoti, izraisa dažādus Šarko-Marijas-Tota slimības apakštipus. Piemēram, RAB7 gēna mutācijas noved pie 2B tipa CMT; SH3TC2 gēna (kas kodē vienu no Švana šūnu membrānas proteīniem) izmaiņas izraisa 4C tipa CMT, kas izpaužas bērnībā un kam raksturīga motoro un sensoro neironu demielinizācija (pastāv pusotrs ducis šīs slimības 4. tipa formu).
Reta 3. tipa CMT (saukta par Dejerine-Sottas sindromu) sāk attīstīties agrā bērnībā, un to izraisa mutācijas PMP22, MPZ, EGR2 un citos gēnos.
Kad 5. tipa CMT rodas 5–12 gadu vecumā, tiek novērota ne tikai motorā neiropātija (apakšējo ekstremitāšu spastiskas paraparēzes veidā), bet arī redzes un dzirdes nervu bojājumi.
Muskuļu vājums un redzes nerva atrofija (ar redzes zudumu), kā arī līdzsvara problēmas ir raksturīgas 6. tipa CMT. Savukārt 7. tipa Šarko-Marijas-Tootas slimības gadījumā ir ne tikai motoriski sensorā neiropātija, bet arī tīklenes slimība pigmentozas retinīta veidā.
Biežāk vīriešiem sastopama ar X hromosomu saistīta KMT jeb Šarko-Marijas-Tootas slimība ar ekstremitāšu tetraparēzi (abu roku un kāju kustību vājums) ir demielinizējošs veids, un tiek uzskatīts, ka to izraisa GJB1 gēna mutācija X hromosomas garajā rokā, kas kodē koneksīnu 32, Švana šūnu un oligodendrocītu transmembrānas proteīnu, kas regulē nervu signālu pārraidi. [ 3 ]
Riska faktori
Galvenais CMT riska faktors ir slimības ģimenes anamnēze, tas ir, tuviem radiniekiem.
Saskaņā ar ģenētiķu datiem, ja abi vecāki ir autosomāli recesīvā Šarko-Marijas-Tootas slimības gēna nesēji, risks, ka bērnam attīstīsies šī slimība, ir 25%. Savukārt risks, ka bērns būs šī gēna nesējs (bet viņam nebūs nekādu simptomu), tiek lēsts 50%.
Ar X hromosomu saistītas iedzimtības gadījumā (kad mutētais gēns atrodas sievietes X hromosomā) pastāv 50% risks, ka māte nodos gēnu savam dēlam, kuram attīstīsies KMT. Slimība var nerasties, piedzimstot meitenei, bet meitas dēli (mazbērni) var mantot bojāto gēnu, un slimība attīstīsies.
Pathogenesis
Jebkura veida Šarko-Marī-Toota slimības gadījumā tās patogenēzi izraisa perifēro nervu iedzimta anomālija: motora (kustību) un sensora (jutīga).
Ja CMT tips ir demielinizējošs, tad mielīna apvalka, kas aizsargā perifēro nervu aksonus, iznīcināšana vai defekts noved pie nervu impulsu pārraides palēnināšanās perifērajā nervu sistēmā - starp smadzenēm, muskuļiem un maņu orgāniem.
Aksonālā slimības tipa gadījumā tiek ietekmēti paši aksoni, kas negatīvi ietekmē nervu signālu stiprumu, kas nav pietiekams pilnīgai muskuļu un maņu orgānu stimulēšanai.
Lasiet arī:
Kā tiek pārnests Šarko-Marijas-Tota sindroms? Bojātos gēnus var mantot autosomāli dominējošā vai autosomāli recesīvā veidā.
Visizplatītākais veids, autosomāli dominējošā iedzimtība, rodas, ja ir viena mutētā gēna kopija (ko nes viens no vecākiem). Un CMT nodošanas varbūtība katram dzimušajam bērnam tiek lēsta 50% apmērā [ 4 ].
Ar autosomāli recesīvu iedzimtību slimības attīstībai nepieciešamas divas bojātā gēna kopijas (pa vienai no katra vecāka, kuram nav nekādu slimības pazīmju).
40–50 % gadījumu rodas autosomāli dominējoša iedzimta demielinizācija, t. i., 1. tipa CMT; 12–26 % gadījumu — aksonāla CMT, t. i., 2. tips. Savukārt 10–15 % gadījumu tiek novērota ar X hromosomu saistīta iedzimtība. [ 5 ]
Simptomi Šarko Marijas-Toota slimība
Parasti pirmās šīs slimības pazīmes sāk parādīties bērnībā un pusaudža gados un pakāpeniski attīstās visas dzīves laikā, lai gan sindroms var izpausties arī vēlāk. Simptomu kombinācija ir mainīga, un slimības progresēšanas ātrumu, kā arī tās smagumu nav iespējams paredzēt.
Tipiski sākotnējās stadijas simptomi ir paaugstināts vispārējs nogurums; samazināts pēdu, potīšu un apakšstilbu muskuļu tonuss (vājums); refleksu trūkums. Tas apgrūtina pēdu kustības un noved pie disbāzes (gaitas traucējumiem), kas izpaužas kā augstāka kāju pacelšanās, bieži vien ar biežiem paklupieniem un kritieniem. Šarko-Marijas-Touta slimības pazīmes mazam bērnam var būt izteikta neveiklība un vecumam neraksturīgas iešanas grūtības, kas saistītas ar divpusēju pēdas noslīdējumu. Raksturīgas ir arī pēdu deformācijas: augsta velve (iedobta pēda) vai izteikta plakanā pēda, izliekti (āmurveida) pirksti.
Ja staigā uz pirkstgaliem muskuļu hipotonijas fonā, neirologs var aizdomas, ka bērnam ir 4. tipa CMT, kurā bērni pusaudža gados var nespēt staigāt.
Slimībai progresējot, muskuļu atrofija un vājums izplatās uz augšējām ekstremitātēm, apgrūtinot smalko motoriku veikšanu un normālus roku uzdevumus. Samazinātas taustes sajūtas un spēja sajust siltumu un aukstumu, kā arī nejutīgums pēdās un rokās norāda uz maņu nervu aksonu bojājumiem.
Šarko-Marī-Toota slimības 3. un 6. tipa gadījumā, kas izpaužas bērnībā, tiek novērota sensoriska ataksija (kustību koordinācijas un līdzsvara traucējumi), muskuļu raustīšanās un trīce, sejas nerva bojājumi, redzes nerva atrofija ar nistagmu un dzirdes zudums.
Vēlākajos posmos var būt nekontrolējama trīce (trīce) un biežas muskuļu krampji; kustību problēmas var izraisīt sāpju attīstību: muskuļu, locītavu, neiropātiskas.
Komplikācijas un sekas
Šarko-Marī-Toota slimībai var būt tādas komplikācijas un sekas kā:
- biežāki sastiepumi un lūzumi;
- kontraktūras, kas saistītas ar periartikulāro muskuļu un cīpslu saīsināšanos;
- skolioze (mugurkaula izliekums);
- elpošanas problēmas – kad tiek bojātas nervu šķiedras, kas inervē diafragmas muskuļus:
- patstāvīgas pārvietošanās spējas zudums.
Diagnostika Šarko Marijas-Toota slimība
Diagnoze ietver klīnisko izmeklēšanu, anamnēzi (ieskaitot ģimenes anamnēzi), neiroloģisko un sistēmisko izmeklēšanu.
Tiek veiktas pārbaudes, lai pārbaudītu kustību diapazonu, jutīgumu un cīpslu refleksus. Nervu vadītspēju var novērtēt, izmantojot instrumentālu diagnostiku – elektromiogrāfiju vai elektroneiromiogrāfiju. Var būt nepieciešama arī ultraskaņa vai MRI. [ 6 ]
Ģenētiskās vai DNS pārbaudes, lai noteiktu visbiežāk sastopamās ģenētiskās mutācijas, kas izraisa CMT asins paraugā, ir ierobežotas, jo DNS testi pašlaik nav pieejami visiem slimības veidiem. Plašāku informāciju skatiet sadaļā Ģenētiskā testēšana.
Dažos gadījumos tiek veikta perifērā nerva (parasti surālā nerva) biopsija.
Diferenciālā diagnoze
Diferenciāldiagnoze ietver citas perifēras neiropātijas, Dišēna muskuļu distrofiju, mielopātiskos un miastēniskos sindromus, diabētisko neiropātiju, mielopātijas multiplās un amiotrofiskās laterālās sklerozes gadījumā, Gijēna-Barē sindromu, peroneālā nerva traumu un tā atrofiju (ieskaitot saspiešanu starp mugurkaula jostas diskiem), smadzenīšu vai talāma bojājumus, kā arī ķīmijterapijas blakusparādības (ārstēšanas laikā ar citostatiskiem līdzekļiem, piemēram, vinkristīnu vai paklitakselu). [ 7 ]
Kurš sazināties?
Profilakse
CMT profilakse var būt nākamo vecāku ģenētiskā konsultēšana, īpaši, ja kādam no pāra ir šī slimība ģimenes anamnēzē. Tomēr ir konstatēti de novo punktu gēnu mutāciju gadījumi, tas ir, ja slimības ģimenes anamnēzē nav.
Grūtniecības laikā Šarko-Marī-Toota slimības iespējamību nedzimušajam bērnam var pārbaudīt ar horiona villus biopsiju (no 10 līdz 13 grūtniecības nedēļām), kā arī ar augļūdeņu analīzi (15-18 nedēļās).
Prognoze
Kopumā dažādu Šarko-Marī-Toota slimības veidu prognoze ir atkarīga no klīniskās smaguma pakāpes, taču visos gadījumos slimība progresē lēni. Daudziem pacientiem ir invaliditāte, lai gan tas nesamazina paredzamo dzīves ilgumu.