
Visi iLive saturs ir medicīniski pārskatīts vai pārbaudīts, lai nodrošinātu pēc iespējas lielāku faktisko precizitāti.
Mums ir stingras iegādes vadlīnijas un tikai saikne ar cienījamiem mediju portāliem, akadēmiskām pētniecības iestādēm un, ja vien iespējams, medicīniski salīdzinošiem pārskatiem. Ņemiet vērā, ka iekavās ([1], [2] uc) esošie numuri ir klikšķi uz šīm studijām.
Ja uzskatāt, ka kāds no mūsu saturiem ir neprecīzs, novecojis vai citādi apšaubāms, lūdzu, atlasiet to un nospiediet Ctrl + Enter.
Treisera Kolinsa sindroms
Raksta medicīnas eksperts

Pēdējā pārskatīšana: 04.07.2025

Intrauterīnie traucējumi kaulu attīstības procesos izraisa nopietnas galvaskausa un sejas deformācijas, un viena no šādas patoloģijas šķirnēm ir Treacher Collins sindroms (TCS) vai mandibulofascial, tas ir, žokļu un sejas disostoze.
Slimības kods saskaņā ar ICD 10: XVII klase (iedzimtas anomālijas, deformācijas un hromosomu traucējumi), Q75.4 - mandibulofaciālā disostoze.
Cēloņi Treisera Kolinsa sindroms
Šis sindroms tika nosaukts izcilā britu oftalmologa Edvarda Trīčera Kolinsa vārdā, kurš pirms vairāk nekā simts gadiem aprakstīja patoloģijas galvenās iezīmes. Tomēr Eiropas ārsti biežāk šāda veida sejas un žokļa kaulu anomāliju sauc par Frančeti slimību vai sindromu – pamatojoties uz Šveices oftalmologa Ādolfasciālās disostozes plašajiem pētījumiem, kurš pagājušā gadsimta vidū ieviesa terminu "mandibulofasciālā disostoze". Medicīnas aprindās tiek lietots arī nosaukums Frančeti-Kolinsa sindroms.
Treačera Kolinsa sindromu izraisa mutācijas TCOF1 gēnā (5q31.3-33.3 hromosomas lokusā), kas kodē nukleolāru fosfoproteīnu, kas ir atbildīgs par cilvēka embrija kraniofaciālās daļas veidošanos. Šī proteīna daudzuma priekšlaicīgas samazināšanās rezultātā tiek traucēta rRNS bioģenēze un funkcijas. Saskaņā ar Cilvēka genoma pētījumu programmas ģenētiķu teikto, šie procesi noved pie neirālā cekuluma - izciļņa gar neirālo rievu, kas embrionālās attīstības laikā aizveras neirālajā caurulītē - embriju šūnu proliferācijas samazināšanās.
Sejas audu veidošanās notiek neirālā cekulu augšējās (galvas) daļas šūnu transformācijas un diferenciācijas dēļ, kuras migrē pa neirālo caurulīti uz embrija pirmā un otrā zaru loka zonu. Šo šūnu deficīts izraisa galvaskausa un sejas deformācijas. Kritiskais periods anomāliju rašanās brīdim ir no 18 līdz 28 dienām pēc apaugļošanās. Pēc neirālā cekulu šūnu migrācijas pabeigšanas (ceturtajā grūtniecības nedēļā) veidojas gandrīz visi irdenie mezenhimālie audi sejas zonā, kas vēlāk (no 5 līdz 8 nedēļām) diferencējas par visu sejas, kakla, balsenes, auss (ieskaitot iekšējo ausi) un topošo zobu skeleta un saistaudiem.
Pathogenesis
Treacher Collins sindroma patoģenēze bieži ir ģimenes, un anomālija tiek mantota autosomāli dominējošā veidā, lai gan ir gadījumi, kad defekts tiek pārmantots autosomāli recesīvi (ar mutācijām citos gēnos, īpaši POLR1C un POLR1D). Visneparedzamākais žokļu un sejas disostozes aspekts ir tas, ka mutācija bērniem tiek mantota tikai 40–48% gadījumu. Tas ir, 52–60% pacientu Treacher Collins sindroma cēloņi nav saistīti ar anomālijas klātbūtni ģimenē, un tiek uzskatīts, ka patoloģija rodas sporādisku gēnu mutāciju rezultātā de novo. Visticamāk, jaunas mutācijas ir teratogēnas ietekmes uz augli sekas grūtniecības laikā.
Starp šī sindroma teratogēnajiem cēloņiem eksperti min lielas etanola (etilspirta) devas, radiāciju, cigarešu dūmus, citomegavīrusu un toksoplazmu, kā arī uz glifosāta bāzes ražotus herbicīdus (Roundal, Glyfor, Tornado u.c.). Un jatrogēno faktoru sarakstā ir aknes un seborejas zāles ar 13-cis-retīnskābi (izotretinoīns, akutāns); pretkrampju līdzeklis fenitoīns (Dilantin, Epanutin); psihotropās zāles diazepāms, valiums, relanijs, seduksens.
Simptomi Treisera Kolinsa sindroms
Lielākoties mandibulofasciālās disostozes klīniskās pazīmes un to izpausmes pakāpe ir atkarīga no gēnu mutāciju izpausmes īpašībām. Un pirmās šīs anomālijas pazīmes vairumā gadījumu ir redzamas bērnam tūlīt pēc piedzimšanas: sejai ar Treacher Collins sindromu ir raksturīgs izskats. Turklāt morfoloģiskās anomālijas parasti ir divpusējas un simetriskas.
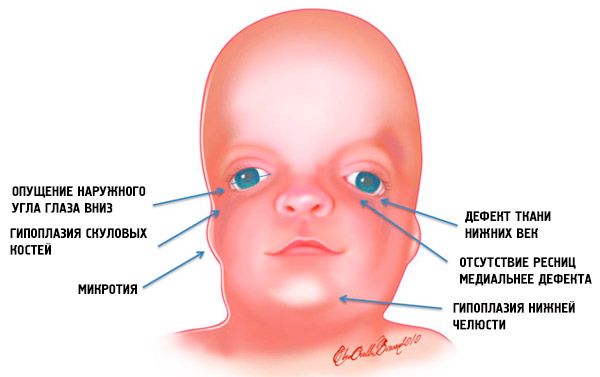
Acīmredzamākie Treacher Collins sindroma simptomi ir:
- galvaskausa sejas kaulu nepietiekama attīstība (hipoplāzija): frontālā kaula zigomatiskie, zigomatiskie izaugumi, sānu pterigoīdās plāksnes, deguna blakusdobumi, apakšžoklis un kaulu epifīžu (kondilu) izvirzījumi;
- apakšžokļa kaulu nepietiekama attīstība (mikrognātija) un apakšžokļa leņķis, kas ir neass nekā parasti;
- deguns ir normāla izmēra, bet šķiet liels uzacu arku hipoplāzijas un vaigu arku nepietiekamas attīstības vai neesamības dēļ deniņu rajonā;
- acu spraugas ir vērstas uz leju, tas ir, acu forma ir patoloģiska, ārējiem kaktiņiem nolaižoties uz leju;
- apakšējo plakstiņu defekti (koloboma) un daļēja skropstu neesamība uz tiem;
- neregulāras formas ausu kaktiņi ar plašu noviržu diapazonu, tostarp to atrašanās vietu apakšžokļa stūrī, daivu neesamību, aklas fistulas starp auss tragusu un mutes kaktiņu utt.;
- ārējās dzirdes kanāla sašaurināšanās vai slēgšanās (atrēzija) un vidusauss kauliņu anomālijas;
- pieauss siekalu dziedzeru neesamība vai hipoplāzija;
- rīkles hipoplāzija (rīkles un elpceļu sašaurināšanās);
- cieto aukslēju nesaplūšana (aukslēju šķeltne), kā arī mīksto aukslēju neesamība, saīsināšanās vai nekustīgums.
Šādām anatomiskām anomālijām visos gadījumos ir komplikācijas. Tie ir funkcionāli dzirdes traucējumi vadītspējas dzirdes zuduma vai pilnīgas kurlības veidā; redzes traucējumi nepareizas acs ābolu veidošanās dēļ; aukslēju defekti, kas rada grūtības baroties un rīt. Pastāv zobu sakodiena traucējumi (maloklūzija), kas saistīti ar žokļa defektiem, kas savukārt rada problēmas ar košļāšanu un artikulāciju. Mīksto aukslēju patoloģijas izskaidro deguna balsi.
Komplikācijas un sekas
Treacher Collins sindroma žokļu un sejas anomāliju sekas ir tādas, ka piedzimstot bērna intelektuālās spējas ir normālas, bet dzirdes defektu un citu traucējumu dēļ tiek novērota sekundāra garīga atpalicība.
Turklāt bērni ar šādiem defektiem akūti izjūt savu mazvērtību un cieš, kas negatīvi ietekmē viņu nervu sistēmu un psihi.
Diagnostika Treisera Kolinsa sindroms
Treacher Collins sindroma pēcdzemdību diagnoze būtībā balstās uz klīniskajām pazīmēm. Kraniofaciālo disostozi ir viegli identificēt, ja sindroms ir pilnībā izteikts, bet, ja ir minimāli izteikti patoloģijas simptomi, var rasties problēmas ar pareizas diagnozes noteikšanu.
Šajā gadījumā īpaša uzmanība jāpievērš visu ar anomālijām saistīto funkciju novērtēšanai, īpaši tām, kas ietekmē elpošanu (miega apnojas riska dēļ). Jānovērtē un jāuzrauga arī barošanas efektivitāte un hemoglobīna skābekļa piesātinājums.
Vēlāk, 5.–6. dienā pēc dzimšanas, dzirdes bojājumu apmērs būs jānosaka, izmantojot audioloģisko pārbaudi, kas jāveic dzemdību namā.
Tiek nozīmēta pārbaude, kuras laikā tiek veikta instrumentālā diagnostika, izmantojot galvaskausa un sejas dismorfoloģijas fluoroskopiju; pantomogrāfija (sejas galvaskausa kaulu struktūru panorāmas rentgenogrāfija); pilna galvaskausa datortomogrāfija dažādās projekcijās; smadzeņu CT vai MRI, lai noteiktu iekšējā dzirdes kanāla stāvokli.
Agrākā – pirmsdzemdību – žokļu un sejas anomāliju diagnoze, ja ģimenes anamnēzē ir Treacher Collins sindroms, ir iespējama ar horiona bārkstiņu biopsiju 10.–11. grūtniecības nedēļā (procedūra apdraud spontāno abortu un dzemdes infekciju).
Asins analīzes tiek veiktas arī no ģimenes locekļiem; 16.–17. grūtniecības nedēļā tiek analizēts amnija šķidrums (transabdominālā amniocentēze); 18.–20. grūtniecības nedēļā tiek veikta fetoskopija un asinis tiek ņemtas no placentas augļa asinsvadiem.
Bet visbiežāk ultraskaņu izmanto šī sindroma pirmsdzemdību diagnostikā auglim (20-24 grūtniecības nedēļās).
Kādi testi ir vajadzīgi?
Diferenciālā diagnoze
Šīs pašas metodes speciālisti izmanto, ja nepieciešama diferenciāldiagnostika, lai atpazītu vieglu Treacher Collins sindromu un atšķirtu to no citām iedzimtām galvaskausa kaulu anomālijām, jo īpaši: Apert, Crouzon, Nager, Peters-Hewels, Hellermann-Steph sindromiem, kā arī hemifaciālas mikrosomijas (Goldenhar sindroms), hipertelorisma, priekšlaicīgas galvaskausa šuvju saplūšanas (kraniosinostoze) vai sejas kaulu saplūšanas traucējumu (kraniosinostoze).
Prognoze
Kāda ir šīs patoloģijas prognoze? Tā ir atkarīga no deformācijas pakāpes un simptomu intensitātes. Trīčera Kolinsa sindroms ir diagnoze, kas jāgaida visu mūžu.
 [ 25 ]
[ 25 ]