
Visi iLive saturs ir medicīniski pārskatīts vai pārbaudīts, lai nodrošinātu pēc iespējas lielāku faktisko precizitāti.
Mums ir stingras iegādes vadlīnijas un tikai saikne ar cienījamiem mediju portāliem, akadēmiskām pētniecības iestādēm un, ja vien iespējams, medicīniski salīdzinošiem pārskatiem. Ņemiet vērā, ka iekavās ([1], [2] uc) esošie numuri ir klikšķi uz šīm studijām.
Ja uzskatāt, ka kāds no mūsu saturiem ir neprecīzs, novecojis vai citādi apšaubāms, lūdzu, atlasiet to un nospiediet Ctrl + Enter.
Smadzeņu lizencefālija
Raksta medicīnas eksperts

Pēdējā pārskatīšana: 12.07.2025
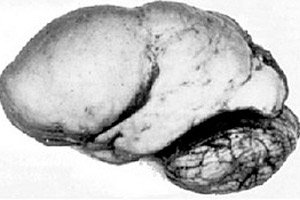
Starp organiskām smadzeņu patoloģijām izceļas tāda iedzimta smadzeņu attīstības anomālija kā lisencefālija, kuras būtība slēpjas gandrīz gludā pusložu garozas virsmā – ar nepietiekamu konvolūciju un vagu skaitu. [ 1 ]
Pilnīgas konvolūcijas neesamības gadījumā tiek definēta agyrija, un vairāku platu, plakanu konvolūciju klātbūtni sauc par pahigiriju. Šiem defektiem, tāpat kā dažām citām smadzeņu redukcijas deformācijām, ICD-10 ir kods Q04.3.
Epidemioloģija
Saskaņā ar reto slimību statistiku, uz 100 tūkstošiem jaundzimušo ir 1–1,2 lisencefālijas gadījumi. [ 2 ], [ 3 ]
Saskaņā ar dažiem datiem, bērniem ar Millera-Dīkera sindromu tiek novēroti līdz pat 25–30 % klasiskās lisencefālijas gadījumu; gandrīz 85 % pacientu tiek atklātas LIS1 un DCX gēnu punktmutācijas un delēcijas. [ 4 ]
17 ar lissencepāliju saistītu gēnu ģenētiskie pētījumi parādīja, ka LIS1 mutācija vai delēcija ir saistīta ar 40% pacientu un 23% ir saistīti ar DCX mutāciju, kam seko TUBA1A (5%) un DYNC1H1 (3%).[ 5 ]
Cēloņi lissencephaly
Visi zināmie smadzeņu garozas (cortex cerebri) veidošanās cēloņi gandrīz vai pilnībā bez līkumiem un vagām, kas palielina cilvēka smadzeņu "darba laukumu" un nodrošina centrālās nervu sistēmas "produktivitāti", ir saistīti ar traucējumiem tās perinatālajā attīstībā. Tas ir, auglim attīstās lisencefālija. [ 6 ]
Augļa smadzeņu garozas slāņu veidošanās neveiksme lissencephalijā ir neironu, kas to veido, patoloģiskas migrācijas vai šī procesa priekšlaicīgas pārtraukšanas rezultāts.
Šis process, kas ir būtisks cerebrokortikālajai histoģenēzei, notiek vairākos posmos no 7. līdz 18. grūtniecības nedēļai. Un, ņemot vērā tā paaugstināto jutību pret ģenētiskām mutācijām, kā arī dažādām negatīvām fizikālām, ķīmiskām un bioloģiskām ietekmēm, jebkura novirze no normas var izraisīt nepareizu neironu lokalizāciju ar iespējamu sabiezējuša pelēkās vielas slāņa veidošanos garozā bez raksturīgas struktūras. [ 7 ]
Dažos gadījumos bērniem lissencephaly ir saistīta ar Miller-Dieker, Walker-Warburg vai Norman-Roberts sindromiem.
Lasiet arī – Smadzeņu attīstības defekti
Riska faktori
Papildus mutācijām dažos gēnos, bērna ar tik smagu defektu piedzimšanas riska faktori ir augļa skābekļa bads (hipoksija); nepietiekama asins piegāde smadzenēm (hipoperfūzija); akūts cerebrovaskulārs negadījums perinatāla insulta veidā; placentas patoloģijas; grūtnieces vīrusu infekcijas (tostarp TORCH); [ 8 ] problēmas ar vispārējo vielmaiņu un vairogdziedzera darbību; smēķēšana, alkohols, psihotropās un narkotiskās vielas; vairāku medikamentu lietošana; paaugstināts radiācijas līmenis. [ 9 ]
Pathogenesis
Ne visiem lissencepālijas gadījumiem patogenezi izraisa hromosomu anomālijas un gēnu mutācijas. Taču ir zināmi daži gēni, kas kodē olbaltumvielas, kurām ir svarīga loma neiroblastu un neironu pareizā kustībā pa radiālajām glijas šūnām, veidojot smadzeņu garozu. Un šo gēnu mutācijas noved pie šīs patoloģijas. [ 10 ]
Konkrēti, tās ir sporādiskas LIS1 gēna mutācijas (bez iedzimtības) 17. hromosomā, kas regulē mikrotubulu citoplazmatisko motoro proteīnu dineīnu, kā arī DCX gēna mutācijas X hromosomā, kas kodē proteīnu dubultkortīnu (lissencephalīnu-X). [ 11 ]. Pirmajā gadījumā speciālisti definē klasisko lissencepāliju (I tips), otrajā – ar X hromosomu saistītu. [ 12 ]
Kad tiek dzēsts FLN1 gēns, kas kodē fosfoproteīnu filamīnu 1, virzītas neironu migrācijas process var vispār nesākties, kā rezultātā pilnībā izzūd konvolūcijas (agirija). [ 13 ]
CDK5 gēnā, kas kodē kināzes enzīmu – intracelulārās metabolisma katalizatoru, regulē šūnu ciklu CNS neironos un nodrošina to normālu migrāciju smadzeņu struktūru pirmsdzemdību veidošanās laikā, ir identificētas mutācijas.
Patoloģiskas izmaiņas RELN gēnā 7. hromosomā, kas izraisa kortikālus girālus defektus Normana-Roberta sindromā, izraisa ekstracelulārā glikoproteīna reelīna deficītu, kas ir nepieciešams, lai regulētu neironu cilmes šūnu migrāciju un pozicionēšanos cortex cerebri attīstības laikā.[ 14 ], [ 15 ], [ 16 ]
ARX gēns kodē nearistalēnisko homeoboksa proteīnu — transkripcijas faktoru, kam ir svarīga loma priekšsmadzenēs un citos audos.[ 17 ] Bērniem ar ARX mutāciju ir arī citi simptomi, piemēram, trūkstošas smadzeņu daļas (corpus callosum ageneze), patoloģiski dzimumorgāni un smaga epilepsija.[ 18 ],[ 19 ]
Ar lissencepāliju ir saistīti vairāki gēni. Šie gēni ir VLDLR, ACTB, ACTG1, TUBG1, KIF5C, KIF2A un CDK5.[ 20 ]
Citomegalovīruss (CMV) ir saistīts ar lissencepālijas attīstību, ko izraisa samazināta asins piegāde augļa smadzenēm. CMV infekcijas smagums ir atkarīgs no gestācijas vecuma. Agrīna infekcija, visticamāk, izraisīs lissencepāliju, jo neironu migrācija notiek grūtniecības sākumā.[ 21 ]
Turklāt šīs anomālijas rašanās mehānisms ietver nepilnīgu vai vēlāku neironu migrācijas pārtraukšanu no periventrikulārās ģeneratīvās zonas uz smadzeņu garozu. Šādos gadījumos attīstās vai nu nepilnīga lissencepālija, vai pahigirija, kurā veidojas vairākas platas rievas un konvolūcijas (bet lielākā daļa no tām nav klāt).
Simptomi lissencephaly
Pirmās šīs patoloģijas pazīmes (ja nav iepriekš minēto sindromu) var neparādīties tūlīt pēc dzimšanas, bet gan pēc pusotra līdz diviem mēnešiem. Un visbiežāk tiek novēroti šādi lissencepālijas klīniskie simptomi:
- muskuļu hipotonija, bieži vien kombinēta ar spastisku paralīzi;
- krampji un ģeneralizēti toniski-kloniski krampji (opistotonusa veidā);
- smaga garīga atpalicība un augšanas aizture;
- neiroloģisko un motorisko funkciju traucējumi.
Rīšanas problēmas rada grūtības barot bērnu. [ 22 ]
Augsta neiromotorisko traucējumu pakāpe bieži izpaužas kā tetraplēģija – visu ekstremitāšu paralīze. Iespējama roku, pirkstu vai kāju pirkstu deformācija.
Normana-Roberta sindroma ar I tipa lissencepāliju gadījumā novēro kraniofaciālas anomālijas: smagu mikrocefāliju, zemu pieres slīpumu un izvirzītu platu deguna tiltiņu, plaši novietotas acis (hiperterlorisms), žokļu nepietiekamu attīstību (mikrognātija). [ 23 ]
Millera-Dīkera sindromu var raksturot arī ar patoloģiski mazu galvas izmēru ar platu, augstu pieri un īsu degunu, ieplakām deniņos (bitemporālām ieplakām) un zemu novietotām, deformētām ausīm.
Smagu lissencepālijas sindromu raksturo mikrocefālija, acs ābolu izmēra samazināšanās (mikroftalmija), kas apvienota ar tīklenes displāziju, obstruktīvu hidrocefāliju un neesošu vai hipoplastisku smadzeņu zvīņainību.
Komplikācijas un sekas
Starp šīs anomālijas komplikācijām speciālisti min rīšanas funkcijas (disfāgijas) un gastroezofageālā refluksa pārkāpumu; refraktāru (nekontrolētu) epilepsiju; biežas augšējo elpceļu infekcijas; pneimoniju (ieskaitot hronisku aspirāciju).
Zīdaiņiem ar lisencefāliju var būt iedzimtas organiskas sirds problēmas, kas izpaužas kā priekškambaru starpsienas defekts vai sarežģīts sirds defekts ar cianozi (Falo tetraloģija). [ 24 ]
Pēcdzemdību augšanas aiztures sekas vairumā gadījumu izraisa nāvi 24 mēnešu laikā pēc dzimšanas.
Diagnostika lissencephaly
Diagnoze sākas ar bērna fizisku pārbaudi, vecāku slimības vēstures izpēti un grūtniecības un dzemdību vēsturi.
Grūtniecības laikā var būt nepieciešama augļa DNS testēšana bez šūnām, amniocentēze vai horiona bārkstiņu paraugu ņemšana. [ 25 ] Plašāku informāciju skatiet sadaļā – Iedzimtu slimību pirmsdzemdību diagnostika.
Instrumentālā diagnostika tiek izmantota, lai vizualizētu smadzeņu struktūras un novērtētu to funkcijas:
- smadzeņu datortomogrāfija;
- smadzeņu magnētiskās rezonanses attēlveidošana (MRI);
- elektroencefalogramma (EEG). [ 26 ]
Grūtniecības laikā var būt aizdomas par lissencepāliju augļa ultraskaņas izmeklējumā pēc 20-21 nedēļas, ja nav parieto-pakauša un kalcīna rievu un smadzeņu Silvija plaisas anomālijas.
Diferenciālā diagnoze
Tiek veikta diferenciāldiagnostika ar citiem iedzimtu smadzeņu defektu sindromiem.
Pastāv vairāk nekā 20 lissencepālijas veidu, no kuriem lielākā daļa iedalās divās galvenajās kategorijās: klasiskā lissencepālija (1. tips) un bruģakmens lissencepālija (2. tips). Katrai kategorijai ir līdzīgas klīniskās izpausmes, bet atšķirīgas ģenētiskās mutācijas.[ 27 ]
Smadzeņu izmeklējumos I tipa lissencepālijas gadījumā smadzeņu garozā ir četri slāņi, nevis seši, kā tas ir novērojams veseliem pacientiem, savukārt 2. tipa lissencepālijas gadījumā smadzeņu garoza ir dezorganizēta un izskatās kunkuļaina vai mezglaina, jo smadzeņu garozu pilnībā ir pārvietojuši kortikālo neironu kopas, ko atdala gliomezenhimālie audi. Pacientiem bija arī muskuļu un acu patoloģijas.
- Klasiskā lissencepālija (1. tips):
- LIS1: Izolēta lissencepālija un Millera-Dīkera sindroms (lissencepālija, kas saistīta ar sejas dismorfismu). [ 28 ]
- LISX1: DCX gēna mutācija. Salīdzinot ar lissencepāliju, ko izraisa LIS1 mutācijas, DCX garozai ir seši, nevis četri slāņi.
- Izolēta lissencepālija bez citiem zināmiem ģenētiskiem defektiem
- Bruģakmens lissencepālija (2. tips):
- Vokera-Varburga sindroms
- Fukujamas sindroms
- Muskuļu, acu un smadzeņu slimības
- Citus tipus nevar ievietot nevienā no divām iepriekš minētajām grupām:
- LIS2: Normana-Roberta sindroms, līdzīgs I tipa lissencepālijai vai Millera-Dīkera sindromam, bet bez 17. hromosomas delēcijas.
- LIS3
- LISX2
Mikrolisencefālija: tā ir normālas kortikālās locīšanās trūkuma un patoloģiski mazas galvas kombinācija. Bērniem ar regulāru lisencefāliju piedzimstot ir normāla izmēra galvas. Bērniem ar mazu galvas izmēru piedzimstot parasti tiek diagnosticēta mikrolisencefālija.
Ir svarīgi arī atšķirt lissencepāliju un polimikrogīriju, kas ir dažādi smadzeņu attīstības defekti.
Kurš sazināties?
Profilakse
Eksperti iesaka topošajiem vecākiem meklēt ģenētiskās konsultācijas, bet grūtniecēm savlaicīgi reģistrēties pie dzemdību speciālistiem un ginekologiem un veikt visas plānotās pārbaudes.
Prognoze
Bērniem ar lisencefāliju prognoze ir atkarīga no tās pakāpes, taču visbiežāk bērna garīgā attīstība nepārsniedz četru līdz piecu mēnešu līmeni. Un visi bērni ar šo diagnozi cieš no smagiem psihomotoriskiem traucējumiem un grūti ārstējamas epilepsijas. [ 30 ]
Saskaņā ar NINDS (Amerikas Nacionālais neiroloģisko traucējumu un insulta institūts) datiem, maksimālais paredzamais dzīves ilgums cilvēkiem ar lissencepāliju ir aptuveni 10 gadi.