
Visi iLive saturs ir medicīniski pārskatīts vai pārbaudīts, lai nodrošinātu pēc iespējas lielāku faktisko precizitāti.
Mums ir stingras iegādes vadlīnijas un tikai saikne ar cienījamiem mediju portāliem, akadēmiskām pētniecības iestādēm un, ja vien iespējams, medicīniski salīdzinošiem pārskatiem. Ņemiet vērā, ka iekavās ([1], [2] uc) esošie numuri ir klikšķi uz šīm studijām.
Ja uzskatāt, ka kāds no mūsu saturiem ir neprecīzs, novecojis vai citādi apšaubāms, lūdzu, atlasiet to un nospiediet Ctrl + Enter.
Keratoderma: cēloņi, simptomi, diagnoze, ārstēšana
Raksta medicīnas eksperts

Pēdējā pārskatīšana: 07.07.2025
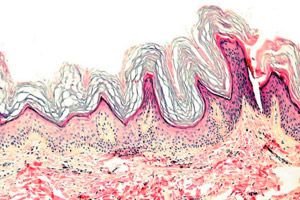
Keratoderma ir dermatožu grupa, kam raksturīgs keratinizācijas procesa traucējums - pārmērīga ragveida audu veidošanās galvenokārt uz plaukstām un pēdām.
Slimības cēloņi un patogeneze nav pilnībā noskaidrota. Pētījumi liecina, ka keratodermas izraisa mutācijas gēnos, kas kodē keratīnu 6, 9, 16. Liela nozīme patoģenēzē ir A vitamīna deficītam, hormonālajiem traucējumiem, galvenokārt dzimumdziedzeru, baktēriju un vīrusu infekcijām. Tās ir viens no iedzimtu slimību un iekšējo orgānu audzēju (parapsoriātiskās keratodermas) simptomiem.
Simptomi. Izšķir difūzo (Unna-Tost keratoderma, Meleda keratoderma, Papillon-Lefevre keratoderma, kropļojošā keratoderma un sindromus, kuros difūzā keratoderma ir viens no galvenajiem simptomiem) un fokālo (izplatīto plankumaino Fišera-Buškes keratodermu, Kosti akrokeratoelastoidozi, Bruhauera-Franzestesta ierobežoto keratodermu, Fuksa lineāro keratodermu u. c.) keratodermu.
Vini-Tosta keratoderma (sinonīmi: iedzimta plaukstu un pēdu ihtioze, Vini-Tosta sindroms) tiek pārmantota autosomāli dominējošā veidā. Pastāv difūza pārmērīga plaukstu un pēdu ādas keratinizācija (dažreiz tikai pēdu), kas attīstās pirmajos divos dzīves gados. Ādas patoloģiskais process sākas ar nelielu plaukstu un pēdu ādas sabiezēšanu bālas krāsas eritēmas sloksnes veidā uz robežas ar veselu ādu. Laika gaitā uz to virsmas parādās gludi, dzeltenīgi ragveida slāņi. Bojājums reti izplatās uz plaukstu locītavu vai pirkstu mugurpusi. Dažiem pacientiem var veidoties virspusējas vai dziļas plaisas, un tiek novērota lokāla hiperhidroze. Autores novērotajam pacientam, tēvocim no mātes puses, brālim un dēlam, bija Vini-Tosta keratoderma.
Aprakstīti nagu (sabiezēšana), zobu un matu bojājumu gadījumi Winy-Tost keratodermā kombinācijā ar dažādām skeleta anomālijām un iekšējo orgānu, nervu un endokrīnās sistēmas patoloģijām.
Histopatoloģija. Histoloģiskā izmeklēšana atklāj izteiktu hiperkeratozi, granulozi, akantozi un nelielus iekaisuma infiltrātus dermas augšējos slāņos. Diferenciāldiagnoze. Slimība jādiferencē no citiem keratodermas veidiem.
Meleda keratoderma (sinonīmi: Meleda slimība, iedzimta progresējoša akrokeratoma, Siemensa plaukstu un pēdu transgradients keratoze, Kogoja iedzimta plaukstu un pēdu progresējoša keratoze) tiek mantota autosomāli recesīvā veidā. Šai keratodermas formai raksturīgi biezi, dzeltenbrūni ragveida slāņi ar dziļām plaisām. Gar bojājuma malām ir redzama vairākus milimetrus plata violeti violeta apmale. Process parasti izplatās uz roku un pēdu aizmuguri, apakšdelmiem un apakšstilbiem. Lielākajai daļai pacientu rodas lokāla hiperhidroze. Šajā sakarā plaukstu un pēdu virsma kļūst nedaudz mitra un pārklāta ar melniem punktiem (sviedru dziedzeru kanāliņiem).
Slimība var attīstīties līdz 15–20 gadu vecumam. Nagi sabiezē un deformējas.
Histopatoloģija. Histoloģiskā izmeklēšana atklāj hiperkeratozi, dažreiz akantozi un hronisku iekaisuma infiltrātu papilārajā dermā.
Diferenciālā diagnoze. Melela keratoderma ir jānošķir no Unna-Tost keratodermas.
Keratoderma Papillon-Lefevre (sinonīms: plaukstu un pēdu hiperkeratoze ar periodontītu) tiek mantota autosomāli recesīvā veidā.
Slimība izpaužas 2.–3. dzīves gadā. Slimības klīniskā aina ir līdzīga Melelas slimībai. Turklāt raksturīgas izmaiņas zobos (piena un pastāvīgo zobu šķilšanās anomālijas ar kariesa attīstību, gingivīts, strauji progresējoša periodontoze ar priekšlaicīgu zobu zaudēšanu).
Histopatoloģija. Histoloģiskā izmeklēšana atklāj visu epidermas slāņu, īpaši ragveida slāņa, sabiezējumu un nenozīmīgus limfocītu un histiocītu šūnu kopumus dermā.
Diferenciālā diagnoze. Slimība jānošķir no citām keratodermām. Svarīga atšķirības pazīme ir raksturīgā zobu patoloģija, kas nav sastopama citās iedzimtas difūzas keratodermas formās.
Keratoderma mutilans (sinonīmi: Fonvinkela sindroms, iedzimta kropļojoša keratoma) ir difūzas keratodermas veids, kas tiek pārmantots autosomāli dominējošā veidā. Tā attīstās 2. dzīves gadā un tai raksturīgi difūzi ragveida nogulumi uz plaukstu un pēdu ādas ar hiperhidrozi. Laika gaitā uz pirkstiem veidojas auklveida rievas, kas noved pie kontraktūrām un pirkstu spontānas amputācijas. Folikulāra keratoze izpaužas roku virspusē, kā arī elkoņa un ceļa locītavu rajonā. Nagu plāksnes ir izmainītas (bieži kā pulksteņa stikliņi). Ir aprakstīti hipogonādisma, rubīna alopēcijas, dzirdes zuduma, pahionihijas gadījumi.
Histopatoloģija. Histoloģiskā izmeklēšana atklāj smagu hiperkeratozi, granulozi, akantozi un nelielus iekaisuma infiltrātus dermā, kas sastāv no limfocītiem un histiocītiem.
Diferenciāldiagnoze. Atšķirot kropļojošo keratodermu no citām difūzās keratodermas formām, vispirms jāņem vērā kropļojošā iedarbība, kas citām formām nav raksturīga. Veicot visu difūzās keratodermas formu diferenciāldiagnostiku, jāatceras, ka tā var būt viens no galvenajiem vairāku iedzimtu sindromu simptomiem.
Ārstēšana. Neotigazons ir indicēts keratodermas vispārējā terapijā. Zāļu deva ir atkarīga no procesa smaguma pakāpes un ir 0,3–1 mg/kg pacienta svara. Neotigazona neesamības gadījumā ieteicams lietot A vitamīnu devā no 100 līdz 300 000 mg dienā ilgstoši. Ārējā terapija sastāv no ziedēm ar aromātiskiem retinoīdiem, keratolītiskiem un steroīdiem līdzekļiem.
 [
[ Kas tevi traucē?
Kas ir jāpārbauda?
Kā pārbaudīt?