
Visi iLive saturs ir medicīniski pārskatīts vai pārbaudīts, lai nodrošinātu pēc iespējas lielāku faktisko precizitāti.
Mums ir stingras iegādes vadlīnijas un tikai saikne ar cienījamiem mediju portāliem, akadēmiskām pētniecības iestādēm un, ja vien iespējams, medicīniski salīdzinošiem pārskatiem. Ņemiet vērā, ka iekavās ([1], [2] uc) esošie numuri ir klikšķi uz šīm studijām.
Ja uzskatāt, ka kāds no mūsu saturiem ir neprecīzs, novecojis vai citādi apšaubāms, lūdzu, atlasiet to un nospiediet Ctrl + Enter.
Angelmana sindroms bērniem un pieaugušajiem
Raksta medicīnas eksperts

Pēdējā pārskatīšana: 04.07.2025

Ir virkne slimību, par kurām tādi izteicieni kā "rūpējies par sevi un tu nesaslimsi", izklausās vismaz smieklīgi. Tās ir patoloģijas, kurās bērna organismā jau pirms dzimšanas ir iedzimtas dažas garīgās un fiziskās anomālijas, taču vecāki par to nav vainīgi. Šādas slimības izraisa mutācijas vai anomālijas hromosomu komplektos, un tās sauc par hromosomu vai ģenētiskām. Angelmana sindroms, Dauna sindroms, Patau sindroms, Edvardsa sindroms, Tērnera sindroms, Prādera-Vilija sindroms - tā ir tikai daļa no ģenētiskajām slimībām no diezgan pieklājīga saraksta.
Laimīga cilvēka sindroms
Šoreiz mēs runāsim par patoloģiju, kas nosaukta angļu pediatra Harija Endželmana vārdā, kurš pirmo reizi aktualizēja šīs problēmas jautājumu 1965. gadā, iepriekšējā dienā savā praksē sastopoties ar trim neparastiem bērniem, kurus vienoja kopīgi savdabīgi simptomi. Ārsts šos bērnus nosauca par leļļu bērniem un uzrakstīja par viņiem rakstu, kas sākotnēji tika nosaukts par "Bērniem-marionešu bērniem". Pats raksts un tā nosaukums tika uzrakstīti, iedvesmojoties no gleznas, kas redzēta vienā no Veronas muzejiem. Gleznā bija attēlots smejošs zēns, un to sauca par "Leļļu zēnu". Gleznā attēlotā bērna saistība ar trim bērniem, kurus Endželmans reiz sastapa savā praksē, pamudināja pediatru apvienot bērnus vienā grupā viņu slimības dēļ.
Nav nekā pārsteidzoša faktā, ka rakstā minētos bērnus citi ārsti nepamanīja. Galu galā no pirmā acu uzmetiena šķita, ka viņiem ir pilnīgi atšķirīgas slimības, tik atšķirīga bija slimības vispārējā klīniskā aina 3 dažādos gadījumos. Varbūt "jaunā" hromosomu patoloģija būtu ieinteresējusi citus zinātniekus, taču tolaik ģenētika vēl nebija pietiekami attīstīta, lai apstiprinātu angļu ārsta hipotēzi. Tāpēc pēc zināmas intereses par to raksts uz ilgu laiku tika nolikts plauktā.
Nākamā Angelmana sindroma pieminēšana, kā tagad sauca angļu pediatra G. Angelmana rakstu, datēta ar 20. gadsimta 80. gadu sākumu. Un tikai 1987. gadā izdevās atrast iemeslu, kāpēc neliela daļa bērnu piedzimst ar tādām novirzēm, ka no malas šķiet, ka viņi pastāvīgi smaida un ir laimīgi. Patiesībā tas nemaz neatbilst patiesībai, un smaids ir tikai grimase, aiz kuras slēpjas nelaimīga cilvēka dvēsele un vecāku sāpes.
Epidemioloģija
Saskaņā ar statistiku, bērna hromosomu mutācija var attīstīties gan uz līdzīgu mutāciju fona vecākiem, gan bez tām. Angelmana sindromam (AS) nav skaidra iedzimta rakstura, taču patoloģijas attīstības varbūtība vecākiem ar hromosomu mutācijām ir diezgan augsta.
Interesanti arī tas, ka, ja ģimenē jau ir bērns ar AS, pastāv viena procenta iespēja, ka otram bērnam būs tāda pati slimība, pat ja vecāki ir veseli.
Joprojām nav precīzas statistikas par Angelmana sindroma pacientu skaitu. Iespējams, iemesls ir simptomu daudzveidība, kas var parādīties noteiktā sastāvā vai ilgstoši nepastāvēt vispār. Tiek pieņemts, ka slimības izplatība ir: 1 bērns uz 20 000 jaundzimušajiem. Taču šis skaitlis ir ļoti aptuvens.
 [
[ Cēloņi Angelmana sindroms
Angelmana sindroms ir medicīnisks nosaukums hromosomu patoloģijai, taču tas nebūt nav vienīgais. Cilvēki šo slimību sauc par leļļu bērnu sindromu, laimīgo leļļu sindromu, Petruškas sindromu un smejošās lelles sindromu. Cilvēki izdomā visādus nosaukumus (dažreiz pat aizskarošus pašiem pacientiem un viņu vecākiem), taču slimība ir slimība, lai cik smieklīga tā neizskatītos un lai kādi būtu tās cēloņi.
Un Angelmana sindroma attīstības iemesli, tāpat kā daudzas citas ģenētiskas patoloģijas, visos gadījumos ir vienas hromosomas vai visa hromosomu komplekta struktūras traucējumi. Bet mūsu gadījumā visa problēma slēpjas 15. hromosomā, kas tiek mantota no mātes. Tas ir, tēva hromosomai šajā gadījumā nav noviržu, bet sieviešu hromosomā notiek noteiktas mutācijas.
Atkarībā no hromosomu anomālijas veida Angelmana sindroms tiek klasificēts kā hromosomu mutācija. Šādas mutācijas tiek uzskatītas par:
- Delēcija (hromosomas daļas, kas satur noteiktu gēnu komplektu, neesamība; ja trūkst viena no gēniem, mēs runājam par mikrodelēciju), kas rodas divu pārrāvumu un vienas atkalapvienošanās rezultātā, kad tiek zaudēta sākotnējās hromosomas daļa.
- Dublēšanās (papildu sadaļas klātbūtne hromosomā, kas ir esošas kopija), kas vairumā gadījumu noved pie cilvēka nāves un retāk pie neauglības.
- Inversija (vienas hromosomas daļas apgriešana par 180 grādiem, t.i., pretējā virzienā, un pēc tam tajā esošie gēni atrodas pretējā secībā), kad hromosomas šķeltie gali ir savienoti secībā, kas atšķiras no sākotnējās.
- Ievietošana (ja daļa no ģenētiskā materiāla hromosomā nav savā vietā),
- translokācija (ja noteikta hromosomas daļa ir piesaistīta citai hromosomai; šāda mutācija var būt savstarpēja, nezaudējot daļas).
Saņemot mutētu hromosomu no neko nenojaušošas mātes, bērns ir lemts piedzimt ar anomālijām. Visbiežākais Angelmana sindroma cēlonis joprojām tiek uzskatīts par mātes 15. hromosomas delēciju, kad trūkst nelielas daļas. Retāk sastopamas mutācijas "smejošās lelles" sindromā ir:
- pārvietošana,
- vienpusēja disomija (ja bērns no tēva saņēmis hromosomu pāri, mātes hromosoma nav),
- DNS gēnu mutācija, kas ir gan galvenais (ģenētiskais) materiāls, gan norādījumi par tā pareizu lietošanu (jo īpaši ube3a gēna mutācija mātes hromosomā).
Vienas no šīm mutācijām klātbūtne vecākiem ir Angelmana sindroma attīstības riska faktors bērniem. Taču ne tikai hromosomu mutācijas, bet arī genomiskās (kas ir saistītas ar kvantitatīvām izmaiņām hromosomu komplektos un ir biežāk sastopamas nekā hromosomu mutācijas) var izraisīt slimības attīstību bērnam. Bieži sastopamas genomiskās mutācijas ir hromosomu trisomija (ja cilvēka hromosomu komplektā ir vairāk nekā 46 hromosomas).
Lai bērnam parādītos patoloģija, vecākiem nemaz nav jābūt hromosomu anomālijām. Un tomēr ir noteikts pacientu procents, kuru slimība ir iedzimta.
Pathogenesis
Iedziļināsimies nedaudz vairāk bioloģijā jeb, precīzāk, ģenētikā. Katra individuālā cilvēka organisma ģenētiskā informācija ir ietverta 23 hromosomu pāros. Viena hromosoma no pāra bērnam tiek nodota no tēva, otra - no mātes. Visi hromosomu pāri atšķiras pēc formas un izmēra un nes noteiktu informāciju. Tādējādi 23. hromosomu pāris (X un Y hromosomas) ir atbildīgs par bērna dzimumpazīmju veidošanos (XX - meitene, XY - zēns, savukārt Y hromosomu bērns var saņemt tikai no tēva).
Ideālā gadījumā bērns no vecākiem saņem 46 hromosomas, kas veido viņa ģenētiskās īpašības, iepriekš nosakot viņu kā indivīdu. Lielāku hromosomu skaitu sauc par trisomiju un uzskata par novirzi no normas. Piemēram, 47. hromosomas klātbūtne hromosomu komplektā (kariotipā, kas nosaka sugu un individuālās īpašības) izraisa Dauna sindroma rašanos.
Ja hromosomas iekrāso ar īpašu krāsvielu, tad mikroskopā gar katru no tām var redzēt dažādu toņu svītras. Katras svītras iekšpusē atrodas milzīgs skaits gēnu. Visas šīs svītras zinātnieki ir numurējuši un tām ir noteikta atrašanās vieta. Vienas svītras neesamība tiek uzskatīta par novirzi no normas. Angelmana sindroma gadījumā ļoti bieži var novērot mātes hromosomas segmentu neesamību intervālā q11-q13, kas atrodas garajā rokā, kurā DNS bāzu skaits ir tikai aptuveni 4 miljoni.
Par hromosomas galveno sastāvdaļu tiek uzskatīta neticami gara DNS molekula, kas satur tūkstošiem gēnu un desmitiem un simtiem miljonu slāpekļa bāzu. Tādējādi 15. hromosoma, kas ir atbildīga par Angelmana sindroma un vairāku citu attīstību, satur 1200 gēnu un aptuveni 100 miljonus bāzu. Jebkādi DNS molekulas struktūras traucējumi noteikti ietekmēs nedzimušā bērna izskatu un attīstību.
Ģenētiskā informācija, kas atrodas gēnos, tiek pārveidota par olbaltumvielām jeb RNS. Šo procesu sauc par gēnu ekspresiju. Tādā veidā no vecākiem saņemtā ģenētiskā informācija iegūst gan formu, gan saturu, kas iemiesojas viņu unikālajā sieviešu vai vīriešu mantiniekā.
Pastāv vairākas patoloģijas ar neklasisku mantojuma veidu, tostarp Angelmana sindroms, kurā gēni, kas saņemti no vecākiem kā daļa no pāra hromosomām, nes unikālu vecāku nospiedumu un izpaužas dažādos veidos.
Tātad, Angelmana sindroms ir spilgts genoma imprintinga piemērs, kurā gēnu ekspresija bērna organismā ir tieši atkarīga no tā, no kura vecāka alēles tika saņemtas (dažādas viena gēna formas, kas saņemtas no tēva un mātes, atrodas identiskās pāru hromosomu daļās). Tas nozīmē, ka tikai anomālijas mātes hromosomā noved pie sindroma attīstības, savukārt tēva hromosomas mutācijas un strukturāli traucējumi izraisa pilnīgi atšķirīgas patoloģijas.

Šajā patoloģijā mātes hromosomā trūkst noteiktu gēnu vai arī atsevišķu gēnu aktivitāte ir samazinājusies/zudusi (lielākajā daļā gadījumu tas ir ube3a gēns, kas ir iesaistīts ubikvitīna, proteīna, kas regulē citu proteīnu noārdīšanos, metabolismā). Tā rezultātā bērnam tiek diagnosticētas garīgās attīstības anomālijas un fiziskas deformācijas.
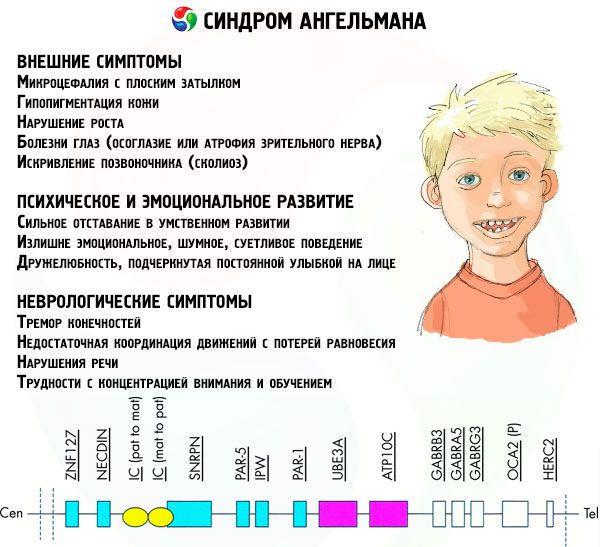
Simptomi Angelmana sindroms
Angelmana sindroma simptomi ietekmē dažādus bērna dzīves un attīstības aspektus: fizisko, neiroloģisko, garīgo. Pamatojoties uz to, var identificēt 3 simptomu grupas, kas norāda uz šīs patoloģijas attīstību.
- Ārējie vai fiziskie simptomi:
- nesamērīgi maza galva salīdzinājumā ar ķermeni un ekstremitātēm, kuru izmērs ir normāls,
- pārāk plata mute,
- gandrīz vienmēr sejā ir smaids (ar atvērtu muti),
- reti zobi,
- šaura augšlūpa,
- bieži izvirzīta plata mēle,
- izvirzīts apakšžoklis,
- smails zods,
- ļoti gaiša āda, bieži mati (albinisms, kas saistīts ar to, ka organisms neražo pigmentu melanīnu),
- tumši plankumi uz gaišas ādas (hipopigmentācija nepietiekamas melanīna ražošanas dēļ)
- fiziski vai ārēji simptomi: acu slimības, piemēram, šķielēšana vai redzes nerva atrofija,
- mugurkaula izliekums (skolioze),
- stīvas kājas (ejot, cilvēks neliec kājas ceļos locītavu zemās kustīguma dēļ, tāpēc salīdzinājums ar lelles gaitu).
- Ar garīgo un emocionālo attīstību saistītie simptomi:
- smaga garīga atpalicība,
- pārāk emocionāla, trokšņaina, niķīga uzvedība,
- bieža roku aplaudēšana,
- izteikta draudzīgums, ko uzsver pastāvīgs smaids sejā,
- bieža smiekli bez iemesla.
- Neiroloģiski simptomi:
- ekstremitāšu trīce,
- nepietiekama kustību koordinācija ar līdzsvara zudumu,
- samazināts muskuļu tonuss,
- dažādi miega traucējumi,
- biežas histēriskas lēkmes bērnībā,
- runas traucējumi (bērns sāk runāt vēlu, viņam ir sliktas komunikācijas prasmes un neskaidra runa),
- hiperaktivitāte uz paaugstinātas uzbudināmības fona,
- grūtības koncentrēties un mācīties.
Bet šis ir vispārināts slimības attēls. Patiesībā Angelmana sindroma klīniskā aina lielā mērā ir atkarīga no slimības attīstības stadijas un hromosomu mutācijas veida, kas izraisīja patoloģiju. Tas nozīmē, ka slimības simptomi dažādiem pacientiem var ievērojami atšķirties, kas ilgu laiku neļāva atšķirt patoloģiju no citiem ar līdzīgu klīnisko ainu.
Starp kopējo simptomu skaitu var izcelt tos, kas raksturīgi visiem pacientiem bez izņēmuma:
- smaga garīga atpalicība,
- nepiemērota uzvedība (nepamatota smiekli, paaugstināta uzbudināmība, slikta koncentrēšanās spēja, eiforijas stāvoklis),
- motorisko prasmju nepietiekama attīstība,
- slikta kustību koordinācija, gaitas ataksija (nevienmērīgs temps, šūpošanās no vienas puses uz otru utt.), ekstremitāšu trīce.
- runas attīstības traucējumi, kuros dominē neverbālie saziņas līdzekļi.
Starp simptomiem, ar kuriem saskaras lielākā daļa pacientu, var atšķirt šādus:
- galvas un ķermeņa nesamērība, ko izraisa aizkavēta fiziskā attīstība,
- daudziem pacientiem galvaskausa forma ir tāda, ka smadzeņu izmērs paliek mazāks nekā veseliem cilvēkiem (mikrocefālija),
- epilepsijas lēkmes pirms 3 gadu vecuma ar pakāpenisku spēka un biežuma samazināšanos vecākā vecumā,
- EEG parametru kropļojumi (zemfrekvences viļņu svārstības un augsta amplitūda).
Šie simptomi ir diezgan izplatīti, tomēr 20% pacientu ar Angelman sindromu to nav.
Vēl retāk ir iespējams diagnosticēt šādas slimības izpausmes kā:
- smags vai viegls šķielēšana,
- slikta mēles kustību kontrole, kā rezultātā pacienti bieži vien bez iemesla izbāž mēli,
- grūtības norīt un sūkt, īpaši maziem bērniem,
- ādas un acu pigmentācijas traucējumi,
- rokas paceltas vai saliektas ejot,
- hiperrefleksija,
- miega traucējumi, īpaši bērnībā,
- bieža siekalošanās,
- neapslāpējamas slāpes,
- pārāk aktīvas košļāšanas kustības,
- paaugstināta jutība pret karstumu,
- plakana galvas aizmugure,
- izvirzīts apakšžoklis,
- gludas plaukstas.
Diezgan lielai daļai pacientu ir problēmas ar urinēšanu, ko viņi slikti kontrolē, traucētas smalkās motorikas, kas rada grūtības pašaprūpē un mācībās, kā arī liekais svars. Gandrīz visiem pacientiem pubertāte iestājas vēlāk nekā veseliem vienaudžiem.
Bērni ar Angelmana sindromu labi uztver mutvārdu runu un saprot to, bet nevēlas piedalīties sarunā, ierobežojot savu runu ar vairākiem desmitiem vārdu, kas nepieciešami ikdienas dzīvē. Tomēr pieaugušā vecumā šādi pacienti izskatās jaunāki par saviem vienaudžiem bez ģenētiskām patoloģijām.
Daudzi Angelmana sindroma simptomi ir nepastāvīgi, tāpēc slimības klīniskā aina ievērojami mainās līdz ar vecumu. Krampji un epilepsijas lēkmes kļūst retākas vai izzūd pavisam, pacients kļūst mazāk uzbudināms un uzlabojas miegs.
Komplikācijas un sekas
Angelmana sindroms ir smaga, pašlaik praktiski neārstējama hromosomu patoloģija, kas liedz pacientiem iespēju dzīvot normālu dzīvi. Kāda būs bērna ar AS dzīve, lielā mērā ir atkarīga no hromosomu anomālijas veida.
Hromosomu segmenta dublēšanās vairumā gadījumu nav savienojama ar dzīvību. Un pat ja šādi pacienti nemirst zīdaiņa vecumā un nesasniedz pubertāti, viņiem nav iespēju radīt bērnus.
Gēnu daļas dzēšana vai neesamība, kas visbiežāk rodas Angelmana sindroma gadījumā, ir šķērslis bērna staigāšanas un runāšanas apguvei. Šādiem bērniem ir smagāka garīgās atpalicības forma, un epilepsijas lēkmes rodas biežāk, un to intensitāte ir daudz lielāka nekā pacientiem ar citām hromosomu anomālijām.
Ja ir tikai viena gēna mutācija, ar pienācīgu uzmanību un pieeju bērnam var iemācīt pašaprūpes, komunikācijas un mijiedarbības pamatus grupā, lai gan attīstībā viņš joprojām atpaliks no vienaudžiem.
Bērniem ar Angelmana sindromu, kuri pēc dabas ir laipni, vissvarīgākā ir vecāku mīlestība un uzmanība. Tikai šajā gadījumā bērna izglītība nesīs augļus, pat ja nelielus. Protams, pacienti ar AS nevarēs mācīties parastā skolā. Viņiem ir nepieciešamas īpašas nodarbības, kur bērniem vispirms iemācīs koncentrēties, un pēc tam pakāpeniski tiks sniegtas skolas zināšanu pamati.
Diagnostika Angelmana sindroms
Angelmana sindroms ir iedzimta attīstības patoloģija. Taču noteiktu apstākļu dēļ to bieži vien nav iespējams diagnosticēt zīdaiņa vecumā un agrā bērnībā. Tas ir saistīts ar simptomu nespecifiskumu un vāju izpausmi zīdaiņiem un bērniem līdz 3 gadu vecumam. Un slimības izplatība mūsu valstī nav tik liela, lai ārsti būtu iemācījušies to atpazīt starp vienaudžiem.
Angelmana sindroms zīdaiņiem var izpausties kā samazināts muskuļu tonuss, kas izpaužas kā problēmas ar barošanos (zūkšanas un rīšanas refleksa vājums), un vēlāk grūtības iemācīties staigāt (šādi bērni sāk staigāt daudz vēlāk). Šie simptomi ir pirmās pazīmes, kas liecina par attīstības anomālijām mazulim, kas var būt saistītas ar hromosomu anomāliju. Tikai ģenētiskā analīze var apstiprināt šo pieņēmumu.
Īpaša uzmanība tiek pievērsta bērniem, kuru vecākiem ir dažādas genoma vai hromosomu anomālijas. Galu galā slimība sākumā var neizpausties, un, ja patoloģija tiek atklāta laikus, sākot intensīvi strādāt ar bērnu, ir iespējams sasniegt ievērojami lielākus panākumus mācībās, palēninot slimības progresēšanu.
Ja vecākiem ir dažādas hromosomu anomālijas, ģenētiskā analīze tiek veikta pat pirms bērna piedzimšanas, jo SA ir viena no patoloģijām, ko var noteikt embrionālajā stadijā.
Materiāla vākšanu ģenētiskajiem pētījumiem var veikt divos veidos:
- invazīvs (ar noteiktu riska procentu, jo ir nepieciešams iekļūt dzemdē, lai ņemtu amnija šķidruma paraugu),
- neinvazīva (bērna DNS analīze no mātes asinīm).
Pēc tam tiek veikti šādi pētījumi:
- Fluorescences in situ hibridizācija (FISH metode) – ar īpašu krāsvielu iezīmētas DNS zondes saistīšana ar pētāmo DNS, kam seko pārbaude mikroskopā.
- ube3a gēna un imprintēto gēnu mutāciju analīze,
- DNS metilēšanas analīze, izmantojot īpašas ģenētikā izmantotas metodes.
Ģenētiskie testi sniedz diezgan precīzu informāciju hromosomu anomāliju gadījumā, kas nozīmē, ka topošie vecāki jau iepriekš zina, kam gatavoties. Tomēr pastāv izņēmumi. Noteiktai pacientu grupai, ja ir visi patoloģiju norādajošie simptomi, testu rezultāti saglabājas normāli. Tas ir, patoloģiju var noteikt, tikai rūpīgi novērojot bērnu jau no agras bērnības: kā viņš ēd, kad sāka staigāt un runāt, vai ejot saliec kājas utt.
Papildus FISH metodei, starp Angelmana sindroma instrumentālajām diagnostikas metodēm var izdalīt tomogrāfiju (CT vai MRI), kas palīdz noteikt smadzeņu stāvokli un lielumu, un elektroencefalogrammu (EEG), kas parāda, kā darbojas atsevišķas smadzeņu daļas.
Ārsti parasti veic galīgo diagnozi 3-7 gadu vecumā, kad pacientam jau ir lielākā daļa simptomu un ir redzama slimības attīstības dinamika.
Kādi testi ir vajadzīgi?
Diferenciālā diagnoze
Angelmana sindroms ir ģenētiska patoloģija, kurai praktiski nav specifisku izpausmju. Lielākā daļa simptomu var vienlīdzīgi norādīt gan uz AS, gan citām ģenētiskām patoloģijām.
Angelmana sindroma diferenciāldiagnoze tiek veikta ar šādām patoloģijām:
- Pita-Hopkinsa sindroms (pacientiem raksturīga garīga atpalicība, dzīvespriecīgs raksturs, smaidoši, viņiem ir diezgan liela un plata mute, tiek atzīmēta mikrocefālija). Atšķirība ir hiperventilācijas un elpas aiztures lēkmes nomoda stāvoklī.
- Kristiansona sindroms (pacienti ir garīgi atpalikuši cilvēki ar dzīvespriecīgu noskaņojumu, nespēj runāt, kam raksturīga mikrocefālija, ataksija, krampji, piespiedu muskuļu kustības).
- Movata-Vilsona sindroms (simptomi: garīga atpalicība, epilepsijas lēkmes, smails zods, atvērta mute, priecīga sejas izteiksme, mikrocefālija). Atšķirības: liels attālums starp acīm, uz iekšu slīpas acis, noapaļots deguna gals, atpakaļ pagriezta auss.
- Kabuki sindroms (kam raksturīga viegla vai vidēji smaga garīga atpalicība, runas un kustību traucējumi, muskuļu vājums, epilepsijas lēkmes, mikrocefālija, gari intervāli starp niezes lēkmēm un koordinācijas traucējumi). Raksturīgas ir izliektas uzacis, apakšējā plakstiņa sānu daļa, plaši novietotas acis, garas plakstiņu plaisas ar garām, biezām skropstām.
- Reta sindroms (diferenciācija no AS sievietēm). Simptomi: aizkavēta runas attīstība, krampji, mikrocefālija. Atšķirība ir tāda, ka sejā nav priecīgas izteiksmes, ir apnojas un apraksijas lēkmes, kas laika gaitā progresē.
- Autosomāli recesīvs garīgās atpalicības sindroms 38 (simptomi: izteikta garīga atpalicība ar aizkavētu motoriku un runas attīstību, muskuļu vājums, barošanās problēmas zīdaiņa vecumā, impulsivitāte). Atšķirīga pazīme ir varavīksnenes zilā krāsa.
- MECP 2 gēna duplikācijas sindroms (diferenciācija no SA vīriešiem). Simptomi: smaga garīga atpalicība, muskuļu vājums kopš bērnības, runas problēmas vai runas trūkums, epilepsija. Atšķirības: progresējoša miopātija, pastāvīgi atkārtotas infekcijas.
- Kleefstras sindroms (simptomi: runas un domāšanas problēmas, muskuļu vājums, miega traucējumi, uzmanības trūkums, atvērta mute, hiperaktivitāte, krampji, ataksija, līdzsvara traucējumi). Raksturīgas pazīmes: plakana seja, īss, strups deguns, plaši novietotas acis, liela, uz iekšu vērsta apakšlūpa, agresīvi uzliesmojumi.
- Smita-Magenisa sindroms (kam raksturīgi krampji, miega problēmas, intelektuālās un motorās attīstības traucējumi). Raksturīgās pazīmes ir plata un plakana seja un izteikta piere.
- Koolen-de-Vries sindroms (viegla vai vidēji smaga garīga atpalicība, muskuļu vājums, krampji, draudzīgums). Atšķirīgas pazīmes: iegarena seja ar augstu pieri, izvirzītas ausis, slīpas acis, augsta locītavu kustība, iedzimti sirds defekti.
- Felana-Makdermida sindroms (simptomi: garīga atpalicība, runas traucējumi vai runas trūkums). Atšķirības: lielas rokas ar attīstītiem muskuļiem, muskuļu vājums kopš dzimšanas, vāja svīšana.
Tādas patoloģijas kā adenilsukcināta deficīts, autosomāli recesīvs garīgās atpalicības sindroms 1, 2q23.1 hromosomas dublēšanās sindroms, FOXG1, STXBP1 vai MEF2C gēna haploinsufficiency sindromi un dažas citas var "lepoties" ar simptomiem, kas līdzīgi Angelmana sindromam.
Ārsta uzdevums ir veikt precīzu diagnozi, diferencējot Angelmana sindromu no patoloģijām ar līdzīgiem simptomiem, un noteikt efektīvu ārstēšanu, kas ir atbilstoša diagnosticētajai slimības stadijai.
Profilakse
Kā lasītājs droši vien jau sapratis, medicīna vēl nespēj novērst gēnu mutācijas un citas hromosomu anomālijas, kā arī situāciju labot. Tas var notikt ar jebkuru, jo bērni ar Angelmana sindromu piedzimst veseliem vecākiem, un ģenētika, kas pašlaik ir viena no vismazāk pētītajām medicīnas nozarēm, to vēl nevar izskaidrot.
Vienīgais, ko var darīt, ir atbildīgi pieiet grūtniecības plānošanai, laikus reģistrēties un veikt pārbaudes. Taču atkal šāds pasākums būs drīzāk izglītojošs, nevis preventīvs, tāpat kā jebkuras pārbaudes. Taču jaunie vecāki jau iepriekš zinās, kam gatavoties, un pozitīvas atbildes gadījumā izlems, vai var uzņemties tādu atbildību kā slima bērna audzināšana.
Prognoze
Angelmana sindroma prognoze ir atkarīga no hromosomu anomālijas rakstura un tās atklāšanas savlaicīguma. Visvairāk cieš tie bērni, kuru 15. hromosomā ir "robi" gēnos (dzēšana). Šādu pacientu staigāšanas un runāšanas varbūtība ir ārkārtīgi zema. Citus gadījumus var labot ar rūpīgu pieeju un mīlestību pret savu bērnu.
Diemžēl šādi pacienti nespēs kļūt par pilnvērtīgiem sabiedrības locekļiem, neskatoties uz to, ka viņi nebūt nav stulbi, viņi saprot runu un tās nozīmi. Tomēr viņiem visu atlikušo mūžu būs problēmas ar komunikāciju. Pacientiem zīmju valodu var mācīt jau no bērnības, taču viņus nevar piespiest sazināties, izmantojot vārdus. "Runājošo" pacientu vārdu krājums ir ierobežots līdz ikdienas dzīvē lietoto vārdu minimumam (5–15 vārdi).
Runājot par Angelmana sindroma pacientu paredzamo dzīves ilgumu un vispārējo veselības stāvokli, šeit skaitļi svārstās ap vidējām vērtībām. Pieaugušā vecumā pacienti galvenokārt saskaras ar tādām veselības problēmām kā skolioze un aptaukošanās, kas ar pareizu ārstēšanas pieeju neapdraud dzīvību.