
Visi iLive saturs ir medicīniski pārskatīts vai pārbaudīts, lai nodrošinātu pēc iespējas lielāku faktisko precizitāti.
Mums ir stingras iegādes vadlīnijas un tikai saikne ar cienījamiem mediju portāliem, akadēmiskām pētniecības iestādēm un, ja vien iespējams, medicīniski salīdzinošiem pārskatiem. Ņemiet vērā, ka iekavās ([1], [2] uc) esošie numuri ir klikšķi uz šīm studijām.
Ja uzskatāt, ka kāds no mūsu saturiem ir neprecīzs, novecojis vai citādi apšaubāms, lūdzu, atlasiet to un nospiediet Ctrl + Enter.
Jauni atklājumi palīdz labāk izprast Retta sindroma cēloņus
Pēdējā pārskatīšana: 02.07.2025

Reta sindroms ir reta neiroloģiskās attīstības slimība, kurai pašlaik nav izārstēšanas vai labas ārstēšanas metodes. Tas izraisa smagus fiziskus un kognitīvus simptomus, no kuriem daudzi pārklājas ar autisma spektra traucējumiem.
Reta sindromu izraisa mutācijas MECP2 gēnā, kas ir ļoti ekspresēts smadzenēs un, šķiet, spēlē svarīgu lomu neironu veselības uzturēšanā. Gēns atrodas X hromosomā, un sindroms galvenokārt skar meitenes. Lai izstrādātu Reta sindroma ārstēšanas metodes, pētnieki vēlas labāk izprast MECP2 un tā funkcijas smadzenēs.
Pētnieki, tostarp Vaithedas institūta līdzdibinātājs Rūdolfs Jaenišs, jau gadu desmitiem pēta MECP2 gēnu, tomēr daudzi pamatfakti par šo gēnu joprojām nav zināmi. Gēna kodētā olbaltumviela MECP2 ir iesaistīta gēnu regulācijā; tā saistās ar DNS un ietekmē dažādu citu gēnu ekspresijas līmeņus jeb to saražotās olbaltumvielas daudzumu.
Tomēr pētniekiem nebija pilnīga MECP2 ietekmēto gēnu saraksta, un nebija vienprātības par to, kā MECP2 ietekmē šos gēnus.
Agrīnie MECP2 pētījumi liecināja, ka tas ir represors, kas samazina mērķa gēnu ekspresiju, taču Jaeniša un citu pētījumi iepriekš bija parādījuši, ka MECP2 darbojas arī kā aktivators, palielinot mērķa gēnu ekspresiju, un ka tas, iespējams, vispār ir aktivators. Tāpat nebija zināms MECP2 darbības mehānisms jeb tas, ko tieši proteīns dara, lai izraisītu izmaiņas gēnu ekspresijā.
Tehnoloģiju ierobežojumi ir liedzuši pētniekiem iegūt skaidrību par šiem jautājumiem. Taču Janišs, viņa laboratorijas pēcdotorants Ji Liu un Janiša bijušais laboratorijas loceklis Entonijs Flamjē, kurš tagad ir docents CHU Sainte-Justine pētniecības centrā Monreālas Universitātē, ir izmantojuši jaunākās metodes, lai atbildētu uz šiem atlikušajiem jautājumiem par MECP2 un iegūtu jaunu ieskatu tā lomā smadzeņu veselībā un slimībās.
Viņu rezultāti tika publicēti žurnālā Neuron, un pētnieki izveidoja arī tiešsaistes MECP2 datu krātuvi — MECP2-NeuroAtlas portālu — kā resursu citiem pētniekiem.
"Es domāju, ka šis raksts fundamentāli mainīs cilvēku izpratni par to, kā MECP2 izraisa Reta sindromu. Mums ir pilnīgi jauna izpratne par mehānismu, un tas varētu sniegt jaunas iespējas slimības ārstēšanas metožu izstrādei," saka Janišs, kurš ir arī bioloģijas profesors MIT.
Dziļāka MECP2 izpratne smadzenēs
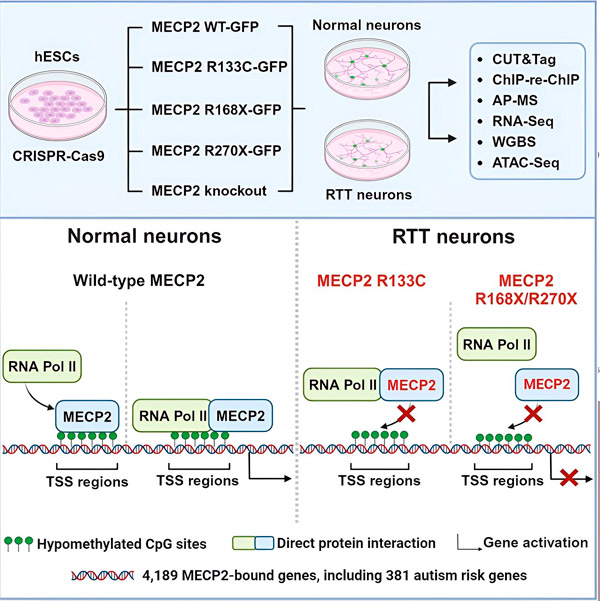
Pētnieki vispirms izveidoja detalizētu karti, kurā parādīts, kur MECP2 saistās cilvēka neironu gēnu sekvencēs, vai nu gēnu iekšienē, vai DNS regulējošajos reģionos to tuvumā. Viņi izmantoja pieeju, ko sauc par CUT&Tag, kas var precīzi noteikt olbaltumvielu mijiedarbību ar DNS.
Pētnieki atrada vairāk nekā 4000 ar MECP2 saistītu gēnu. Viņi atkārtoja savu kartēšanu neironos ar izplatītām MECP2 mutācijām, kas saistītas ar Reta sindromu, lai noteiktu, kur slimības stāvoklī MECP2 ir noplicināts.
Zinot, pie kuriem gēniem saistās MECP2, Liu un Flamjē varēja sākt sasaistīt MECP2 mērķus ar smadzeņu veselību. Viņi atklāja, ka daudzi no tā mērķiem ir iesaistīti neironu aksonu un sinapšu attīstībā un funkcijā.
Viņi arī salīdzināja savu MECP2 mērķu sarakstu ar Simons Foundation Autism Research Initiative (SFARI) datubāzi par autisma izraisītiem gēniem un atklāja, ka 381 gēns šajā datubāzē ir MECP2 mērķi.
Avots: Neuron (2024). DOI: 10.1016/j.neuron.2024.04.007
Šie atklājumi var palīdzēt noskaidrot mehānismus, kas ir autisma simptomu pamatā Reta sindromā, un sniegt labu sākumpunktu MECP2 iespējamās lomas autisma attīstībā izpētei.
"Mēs esam izveidojuši pirmo integrēto MECP2 epigenoma karti veselības un slimību kontekstā, un šī karte var vadīt turpmākos pētījumus," saka Liu. "Zināšanas par to, kuri gēni ir MECP2 mērķi un kuri gēni slimības laikā tiek tieši traucēti, nodrošina stabilu pamatu Reta sindroma izpratnei un jautājumu uzdošanai par gēnu regulāciju neironos."
Pētnieki arī pētīja, vai MECP2 palielināja vai samazināja mērķa gēnu ekspresiju. Saskaņā ar MECP2 vēsturi, ko daži identificēja kā aktivatoru, bet citi - kā represoru, Liu un Flamjērs atrada piemērus, kur MECP2 spēlēja abas lomas.
Tomēr, lai gan MECP2 biežāk tiek uzskatīts par represoru, Liu un Flamjērs atklāja, ka tas galvenokārt ir aktivators, apstiprinot iepriekšējos Jaeniša un Liu atklājumus. Viens jauns eksperiments parādīja, ka MECP2 aktivizē vismaz 80% savu mērķu, bet cits atklāja, ka tas aktivizē līdz pat 88% savu mērķu.
Pētnieku izveidotā mērķa gēnu karte sniedza papildu ieskatu MECP2 lomā kā aktivatoram. Viņi atklāja, ka gēniem, kurus aktivizē MECP2, tas parasti saistās ar DNS reģionu augšpus gēna, ko sauc par transkripcijas sākuma vietu.
Šī ir vieta, kur šūnu mehānisms uzsāk gēna transkripcijas procesu RNS, pēc kura RNS tiek translēta funkcionālā olbaltumvielā, kas ir gēnu ekspresijas produkts. MECP2 klātbūtne transkripcijas sākuma vietā, kur sākas gēnu ekspresija, atbilst tā lomai kā gēnu aktivatoram.
Pēc tam pētnieki sāka pētīt MECP2 lomu gēnu aktivācijā. Viņi pētīja, pie kādām molekulām MECP2 saistās šajā vietā papildus DNS, un atklāja, ka MECP2 tieši mijiedarbojas ar olbaltumvielu kompleksu, ko sauc par RNS polimerāzi II (RNS Pol II). RNS Pol II ir galvenā šūnu mašīna, kas pārraksta DNS RNS. RNS Pol II pati nevar atrast gēnus, tāpēc tai ir nepieciešami dažādi kofaktori jeb olbaltumvielu līdzstrādnieki, lai palīdzētu tai veikt savu darbu.
Pētnieki ierosina, ka MECP2 kalpo kā viens no šādiem kofaktoriem, palīdzot RNS Pol II uzsākt transkripciju gēnos, kur saistās MECP2. MECP2 strukturālā analīze ir identificējusi molekulas daļas, kas saistās ar RNS Pol II, un citi eksperimenti ir apstiprinājuši, ka MECP2 zudums samazina RNS Pol II klātbūtni atbilstošās transkripcijas sākuma vietās, kā arī mērķa gēnu ekspresijas līmeņus.
Tas liek domāt, ka Reta sindromu var izraisīt MECP2 mērķa gēnu transkripcijas samazināšanās MECP2 mutāciju dēļ, kas neļauj tam saistīties ar RNS Pol II vai saistīties ar DNS. Saskaņā ar šo ideju, visbiežāk ar slimībām saistītās MECP2 mutācijas ir saīsinājumi: mutācijas, kurās trūkst daļas proteīna, kas var mainīt MECP2 un RNS Pol II mijiedarbību.
Pētnieki cer, ka viņu atklājumi ne tikai mainīs mūsu izpratni par MECP2, bet arī dziļāka un plašāka izpratne par to, kā MECP2 ietekmē smadzeņu attīstību un funkcijas, varētu novest pie jaunām atziņām, kas palīdzēs cilvēkiem ar Reta sindromu un ar to saistītiem traucējumiem, tostarp autismu.
"Šis projekts ir lielisks piemērs Janiša laboratorijas sadarbības raksturam," saka Flamjērs. "Mums ar Rūdolfu bija īpaša problēma, kas saistīta ar Reta sindromu, un man bija pieredze ar CUT&Tag tehnoloģiju, kas varētu atrisināt šo problēmu. Diskusijas gaitā mēs sapratām, ka varam apvienot savus centienus, un tagad mums ir lieliska informācijas krātuve par MECP2 un tā saistību ar slimībām."