
Visi iLive saturs ir medicīniski pārskatīts vai pārbaudīts, lai nodrošinātu pēc iespējas lielāku faktisko precizitāti.
Mums ir stingras iegādes vadlīnijas un tikai saikne ar cienījamiem mediju portāliem, akadēmiskām pētniecības iestādēm un, ja vien iespējams, medicīniski salīdzinošiem pārskatiem. Ņemiet vērā, ka iekavās ([1], [2] uc) esošie numuri ir klikšķi uz šīm studijām.
Ja uzskatāt, ka kāds no mūsu saturiem ir neprecīzs, novecojis vai citādi apšaubāms, lūdzu, atlasiet to un nospiediet Ctrl + Enter.
Fenilpiruvīna oligofrēnija vai fenilketonūrija
Raksta medicīnas eksperts

Pēdējā pārskatīšana: 05.07.2025
Saskaņā ar statistiku, vienam no 10–15 tūkstošiem jaundzimušo tiek diagnosticēta ģenētiski noteikta neirokognitīvā patoloģija – fenilpiruviskā oligofrēnija, kas attīstās iedzimta neaizvietojamās aminoskābes fenilalanīna metabolisma traucējuma dēļ.
Slimību pirmo reizi identificēja 20. gs. trīsdesmitajos gados Norvēģijā ārsts Ivars Fēlings, kurš to nosauca par hiperfenilalaninēmiju. Tagad šo patoloģiju parasti sauc par fenilketonūriju, tās ICD 10 kods ir E70.0.
 [
[ Fenilpiruviskās oligofrēnijas cēloņi
Fenilpiruviskās oligofrēnijas ģenētiskie cēloņi ir recesīva bērna mantošana no vecākiem, kuri ir divu mutētu enzīma fenilalanīna hidroksilāzes (fenilalanīna-4-monooksigenāzes) alēļu nesēji. Tā rezultātā bērnam pilnībā vai daļēji trūkst šī enzīma, kas ir nepieciešams fenilalanīna (proteinogēnās α-amino-β-fenilpropionskābes) sadalīšanai tirozīnā [2-amino-3-(4-hidroksifenil)propānskābe].
Ģenētiķi ir noskaidrojuši, ka šīs slimības patogeneze ir saistīta ar vairāk nekā 500 dažādām homozigotām vai heterozigotām strukturālām mutācijām 12. hromosomā - aknu enzīma fenilalanīna hidroksilāzes 12q23.2 vai 12q22-q24.1 gēnos. Gēna mutācija noved pie tā, ka šī enzīma līmenis, kas nepieciešams olbaltumvielu pārtikā esošā fenilalanīna oksidēšanai, samazinās 4 vai vairāk reizes.
Tā rezultātā, no vienas puses, asinīs uzkrājas fenilalanīna pārpalikums, kas ir toksisks smadzeņu attīstībai. Šajā gadījumā fenilalanīns, ko nevar pārvērst par tirozīnu, sadalās deaminācijas ceļā, veidojot fenilpiruvskābi (2-okso-3-fenilpropionskābi jeb fenilpiruvātu). Šī skābe savukārt tiek pārvērsta feniletiķskābē (fenilacetātā) un fenilpienskābē (fenilaktātā), kas negatīvi ietekmē smadzeņu un centrālās nervu sistēmas šūnas.
No otras puses, fenilpiruviskās oligofrēnijas galvenie cēloņi (iedzimts fenilalanīna hidroksilāzes deficīts un tā sekas - fenilalanīna pārpalikums) noved pie citu aminoskābju (triptofāna, treonīna, metionīna, valīna, izoleicīna, leicīna) līmeņa pazemināšanās smadzenēs, kas pilnībā izjauc nervu impulsus pārraidošo neirotransmiteru - dopamīna un norepinefrīna - biosintēzes procesu un izraisa progresējošus garīgās un fiziskās attīstības traucējumus bērniem.
Fenilpiruviskās oligofrēnijas simptomi
Zīdaiņiem organismā izpaužas fenilalanīna pārpalikums, un tā pirmās pazīmes ir: letarģija un miegainība; problēmas ar zīšanu; krampji; vemšana; epidermas ekzēmas bojājumi; pelējuma (peles) smaka no ķermeņa, urīna un elpas (paaugstināta fenilacetāta satura dēļ, kas oksidējas fenilketonos).
Tiek novēroti šādi tipiski fenilpiruviskās oligofrēnijas simptomi: augšanas aizkavēšanās; muskuļu hiperkinēze, trīce vai samazināts muskuļu tonuss; psihotiski lēkmes, ko pavada nemotivēta aizkaitināmība un dusmas; samazinātas intelektuālās spējas vai garīga atpalicība.
Bērniem ar fenilketonūriju ir arī hipopigmentācija – ievērojami gaišāka un plānāka ādas, matu un acu krāsa salīdzinājumā ar vecākiem. Šī simptoma tiešais cēlonis ir tirozīna deficīts, kas, oksidējoties epidermas melanocītos, veido pigmentu melanīnu.
Kā atzīmē bērnu psihiatrijas speciālisti, fenilpiruviskās oligofrēnijas klīniskā aina ir atkarīga no smadzeņu šūnu bojājuma pakāpes, un mazulis ar šo patoloģiju pirmajos dzīves mēnešos var šķist normāls. Ja slimība netika atklāta tūlīt pēc piedzimšanas un bērns ilgstoši nesaņēma ārstēšanu, tad sekas ir: neatgriezeniski smadzeņu bojājumi un to komplikācijas smagas vai dziļas garīgās atpalicības (imbecilitātes vai idiotisma) veidā.
Un tad var rasties šādi fenilpiruviskās oligofrēnijas simptomi: īss augums, galvaskausa un sejas anomālijas (mikrocefālija, izvirzīts augšžoklis, plaši izvietoti zobi, traucēta zobu emaljas attīstība), paaugstināts muskuļu tonuss un cīpslu refleksi, hiperaktivitāte, slikta kustību koordinācija un neveikla gaita, paaugstināta aizkaitināmība un citas uzvedības un neiroloģiskas problēmas līdz pat garīgiem traucējumiem.
Fenilpiruviskās oligofrēnijas diagnoze
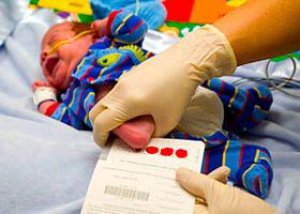
Fenilpiruviskās oligofrēnijas agrīna diagnostika un tūlītēja ārstēšana ir ārkārtīgi svarīga, samazinot garīgās atpalicības attīstības risku no 80–90 % līdz 6–8 % (saskaņā ar CLIMB – Apvienotās Karalistes Nacionālo vielmaiņas slimību informācijas centru).
Šajā nolūkā zīdaiņiem trešajā līdz ceturtajā dienā pēc dzimšanas (izņēmuma gadījumos starp 5. un 8. dienu) jāveic jaundzimušo fenilketonūrijas skrīnings, izmantojot mikrobioloģisku asins analīzi fenilalanīna satura noteikšanai – tā saukto Guthrie testu.
Fenilpiruvāta bioķīmiskās urīna analīzes arī atklāj fenilketonūriju jaundzimušajiem, taču šī analīze jāveic tikai 10–12 dienas pēc dzimšanas. Tāpēc starptautiskais diagnostikas standarts ir fenilalanīna koncentrācijas noteikšana asins plazmā.
Mūsdienīga instrumentāla diagnostika, kuras pamatā ir jaunas tehnoloģijas, piemēram, tandēma masas spektrometrija, ļauj atklāt daudzus iedzimtus vielmaiņas traucējumus, kas izraisa intelektuālās attīstības traucējumus. Tas nodrošina ģenētisko vielmaiņas patoloģiju diferenciāldiagnostiku, tostarp: galaktozēmiju, ketonūriju (leicīna, izoleicīna un valīna metabolisma traucējumi), homocistinūriju, glutārskābes acidūriju, izovalerskābes acidēmiju, sirpjveida šūnu anēmiju, tirozinēmiju, iedzimtu hipotireozi u.c.
Kurš sazināties?